Causas genéticas de Convulsiones Neonatales
Febrero 2021
Etiologías genéticas
de Convulsiones Neonatales
Shagun Kaur Division of Genetic, Genomic, and Metabolic Disorders, Children’s Hospital of Michigan, Detroit, MI
Neoreviews Vol. 21 No. 10 Octubre 2020
Brechas de práctica
Se espera que los
neonatólogos sean capaces de manejar las convulsiones neonatales e
identificar la causa subyacente. Estos médicos deben ser conscientes
de la posibilidad y la probabilidad de las etiologías genéticas de
las convulsiones neonatales en diferentes escenarios clínicos. Los
neonatólogos deben consultar adecuadamente la genética y, junto con
los consultores, ordenar investigaciones y ofrecer terapias
dirigidas.
Objetivos
Identificar cuándo es apropiada una evaluación genética para las convulsiones neonatales
Reconocer diferentes categorías de causas genéticas de convulsiones neonatales.
Ser capaz de evaluar las causas genéticas de las convulsiones neonatales en un enfoque paso a paso.
Reconocer los trastornos convulsivos neonatales y las terapias dirigidas disponibles.
Introducción
Las convulsiones neonatales tienen una tasa de incidencia de alrededor de 3 por cada 1000 nacidos vivos con un inicio que varía desde el nacimiento hasta las 4 semanas de edad (44 semanas de edad posmenstrual). (1) Pueden ocurrir convulsiones aisladas debido a una disfunción cerebral temporal aguda por diversos factores perinatales, pero la epilepsia neonatal se define por la presencia de convulsiones espontáneas recurrentes. Las convulsiones son un motivo común de consulta genética en la UCIN, y de manera apropiada, porque la mayoría de los recién nacidos con epilepsia neonatal tendrán una etiología genética subyacente.
Un estudio reciente en 2017 encontró un diagnóstico genético en 34 (59%) de 58 recién nacidos con epilepsia que se sometieron a pruebas genéticas. (1) (2)
Varias afecciones genéticas pueden causar epilepsia neonatal, incluidos trastornos cromosómicos, de un solo gen y metabólicos. La nueva disponibilidad relativamente generalizada de paneles multigénicos, secuenciación del exoma completo y secuenciación del genoma completo ha aumentado nuestra capacidad para detectar las causas genéticas de las convulsiones. No solo está aumentando nuestro rendimiento diagnóstico, sino que también está aumentando el potencial para el tratamiento dirigido de los trastornos epilépticos neonatales. Aunque nos centraremos en la etiología genética de las convulsiones, se debe considerar un diagnóstico diferencial amplio, como ocurre con cualquier otro proceso diagnóstico; en este caso, el diagnóstico diferencial debe incluir encefalopatía hipoxico isquémica, infección, desequilibrio electrolítico y hemorragia intracraneal.
Tipos de alteraciones convulsivas
neonatales
La epilepsia neonatal
puede ser clasificada en las siguientes categorías :
Malformaciones cerebrales estructurales
Errores congénitos del metabolismo (IEMs);
Sindrómica
No sindrómica, 1 solo gen.
1.- Malformaciones cerebrales
estructurales
Las malformaciones
estructurales aisladas pueden causar convulsiones neonatales debido
a anomalías focales o difusas en el tamaño y la formación del
cerebro, o anomalías de la migración neuronal. Las anomalías
estructurales tales como agenesia del cuerpo calloso (ACC), polimicrogiria,
lisencefalia, esquizencefalia, megalencefalia, holoprosencefalia y las heterotopías de la
materia gris pueden causar convulsiones. De estos defectos, la ACC
es un hallazgo relativamente común (prevalencia ~ 5 / 1000). Aunque
las convulsiones aisladas pueden ser comunes, cuando ocurren como
parte de una secuencia de malformación craneal, hasta 13% de los
pacientes se atribuyen a ACC aislada. (3) Todas las anomalías
estructurales enumeradas en este documento se producen debido a la
alteración de la embriogénesis normal y, a menudo, una mutación en
un solo gen puede provocar malformaciones de múltiples estructuras
en el cerebro. Se requieren neuroimágenes e interpretación
detalladas por parte de un neurorradiólogo pediátrico para una
identificación precisa de la causa. El daño atóxico, tal como una
infección o una deficiencia vascular, al embrión durante las etapas
críticas puede simular anomalías debido a defectos genéticos.
Las anomalías estructurales del cerebro pueden estar aisladas o formar parte de una constelación de características anormales en un síndrome genético. Se debe considerar la vigilancia sindrómica para buscar la participación de otros sistemas u órganos. La Tabla 1 (al final de este artículo) enumera algunas causas genéticas sindrómicas y no sindrómicas comunes de malformaciones cerebrales estructurales. La edad a la que ocurren las convulsiones debido a malformaciones estructurales subyacentes varía mucho, pero en un informe reciente se identificó que ~ 28% de los recién nacidos padecían epilepsia debido a una malformación cerebral. Se encontró que la mayoría de los pacientes con presentación temprana tenían anomalías en la migración neuronal. (1)
2.- Errores congénitos del metabolismo
Los trastornos metabólicos
rara vez son la causa subyacente de un recién nacido que presenta
convulsiones, pero no deben pasarse por alto, ya que algunos se
pueden tratar con cambios en la dieta o medicamentos (tabla 2)
(al final de este artículo) . Los
IEM pueden causar convulsiones debido a varias razones, incluida la
deficiencia de energía, acumulación de productos tóxicos,
anomalías del metabolismo de los neurotransmisores o las
malformaciones cerebrales acompañantes. (4) Las pistas para un IEM
subyacente incluyen la resistencia a terapia con fármacos
antiepilépticos estándar (FAE), características tales como hipotonía o
mala alimentación, o hallazgos de imágenes de resonancia magnética (MRI)
del cerebro que sugieren un trastorno metabólico. (Tabla 3; ejemplo
mostrado en la Figura).
(al final de este artículo) . Otras pistas en la evaluación de laboratorio
incluyen hipoglucemia, acidosis metabólica sin brecha aniónica,
acidosis láctica, cetosis e hiperamonemia.
El enfoque estándar de las
convulsiones, que consiste en establecer el tipo clínico e
identificar anomalías características en la electroencefalografía (EEG),
puede
no ser útil, porque estos hallazgos no son específicos en la
mayoría de los pacientes con IEM. Las convulsiones en los trastornos
metabólicos pueden ser una manifestación primaria o secundaria a
anomalías metabólicas como hipoglucemia o hiperamonemia. La mayoría
de los pacientes con convulsiones en el contexto de un trastorno
metabólico finalmente requerirán un tratamiento con FAE a largo
plazo (fármacos antiepilépticos) . (4)
La presencia de hipoglucemia es un signo de deficiencia energética
y, si está presente, se deben considerar en el diagnóstico diferencial los
siguientes trastornos metabólicos : defectos de gluconeogénesis,
trastornos del almacenamiento de glucógeno, trastornos del
metabolismo de la fructosa, trastornos de la oxidación de ácidos
grasos y trastornos del transporte de glucosa. . Todos los niños con
convulsiones hipoglucémicas deben recibir una evaluación metabólica
que incluya glucosa en sangre, cetonas en suero y orina, ácidos
grasos libres, ácido láctico, aminoácidos plasmáticos,
acilcarnitinas plasmáticas, ácidos orgánicos en orina, amonio,
insulina, hormona del crecimiento y cortisol.
Estas pruebas de laboratorio deben realizarse en el momento de la hipoglucemia para que la interpretación sea más precisa. Las extracciones de sangre tardías pueden permitir que los metabolitos se normalicen, lo que conduce a una interpretación inexacta y diagnósticos errados. Aminoacidopatías, acidurias orgánicas y defectos del ciclo de la urea se presentan con convulsiones cuando hay acumulación de productos tóxicos. El paso de los productos tóxicos a través de la barrera hematoencefálica puede causar edema cerebral, tal como el que se observa en la enfermedad de la orina con jarabe de arce, o neurotoxicidad directa causada por hiperamonemia, tal como en las acidemias orgánicas y los trastornos del ciclo de la urea.
Las convulsiones neonatales
resistentes a los FAE habituales pueden ser el resultado de una de
las epilepsias neonatales que responden a las vitaminas. La
epilepsia dependiente de piridoxina se produce por una deficiencia
de semialdehído alfa-aminoadípica deshidrogenasa (antiquitina) en la
vía de degradación de la lisina. Este bloqueo conduce a la
acumulación de semialdehído alfa-aminoadípico, piperideina-6-carboxilato y ácido pipecólico. Las convulsiones pueden controlarse
con la administración de vitamina B6, pero los retrasos en el
desarrollo pueden persistir. La deficiencia de piridoxamina
59-fosfato oxidasa puede causar convulsiones neonatales e
intrauterinas con hipoglucemia y acidosis láctica. Los estudios de
líquido cefalorraquídeo (LCR) pueden demostrar elevación de glicina, treonina, L-DOPA y 3-metoxitirosina. Debido a que el defecto está en
la conversión de piridoxina a la forma activada de piridoxal fosfato
(PLP), el tratamiento implica la suplementación con PLP. Se ha
sugerido que los recién nacidos prematuros con signos de
encefalopatía hipóxico-isquémica con convulsiones refractarias a los
FAE se sometan a ensayos de PLP. Sin diagnosticar y sin tratar,
estas condiciones pueden tener una morbilidad y mortalidad severas
debido a un estado epiléptico prolongado. (5) Las convulsiones que
responden al ácido folínico son clínicamente indistinguibles de las
epilepsias dependientes de piridoxina y responden al tratamiento con
ácido folínico.
La deficiencia de biotinidasa, si no se diagnostica ni se trata, puede presentarse con convulsiones neonatales graves. Las formas graves de defectos del metabolismo de los ácidos grasos tales como la deficiencia de carnitina-palmitoil transferasa II y las condiciones de acidosis láctica congénita (tales como la deficiencia de piruvato carboxilasa y de piruvato deshidrogenasa) pueden causar cambios quísticos en el cerebro y potencialmente convulsiones. Los trastornos peroxisomales y los trastornos congénitos de la glicosilación también pueden presentarse con convulsiones neonatales y malformaciones del cerebro tales como polimicrogiria frontal y paquigiria. La deficiencia grave de metilentetrahidrofolato reductasa puede causar convulsiones neonatales y se detecta midiendo los niveles de homocisteína. La hiperglicinemia no cetósica puede presentarse con convulsiones progresivas, espasmos inicialmente mioclónicos y elevación aislada de la relación entre el glicina en líquido cefalorraquídeo y glicina plasmática. La acumulación prenatal de glicina puede causar lesión cerebral en útero y movimientos fetales anormales caracterizados como "hipo". Se puede demostrar una disminución de la actividad del sistema de escisión (cleavage) de la glicina en las vellosidades coriónicas y un aumento de glicina en el líquido amniótico.
Otras causas metabólicas de epilepsia, tales como trastornos mitocondriales, trastornos del metabolismo de la creatina, deficiencia del transportador de glucosa tipo 1 y lipofuscinosis ceroide neuronal, típicamente presentes en la infancia o más tarde, pero podrían considerarse en el diferencial de convulsiones neonatales si existen otros aspectos del cuadro clínico que levante sospechas.
Sindrómica
Las epilepsias sindrómicas
suelen tener compromiso multisistémico y también pueden tener rasgos
dismórficos característicos. Trastornos cromosómicos tales como
trisomía 13, 18 o 21; Síndrome de deleción 22q11.2; y síndrome de
Wolf-Hirschhorn se pueden asociar con epilepsia neonatal. Algunos
síndromes genéticos comunes que pueden causar malformaciones
cerebrales estructurales a menudo se presentan con convulsiones
neonatales, como se enumera en la
Tabla 1.
(al final del capítulo) .
Las convulsiones pueden ser el primer síntoma de síndromes neurocutáneos como esclerosis tuberosa, síndrome de Sturge-Weber o incontinencia pigmentaria. Los trastornos vasculares relacionados con COL4A1 pueden tener convulsiones neonatales, ya sea secundarias a hemorragia o como resultado de porencefalia o esquizencefalia subyacente. La epilepsia es una característica de cientos de otros síndromes genéticos y, en muchos, las convulsiones pueden comenzar en la infancia. La Tabla 4 enumera una selección de síndromes genéticos que pueden presentarse con epilepsia neonatal.
1 solo Gen / No sindrómica
Los trastornos de epilepsia
de un solo gen pueden resultar de modificaciones de genes implicados
en la regulación de los canales iónicos, función sináptica o
señalización celular anormal. Las canalopatías pueden ocurrir a
partir de variantes en genes como KCNT1, KCNQ2, CACNA1A o SCN2A, los que
regulan la función de los canales de calcio, potasio o sodio y
juegan un papel en los potenciales de acción y activación
neuronal. A menudo, la expresividad asociada con estos genes es
variable, con rango desde epilepsia neonatal familiar benigna hasta las
encefalopatías epilépticas graves, a veces con asociaciones de
genotipo-fenotipo.
Los canales de cloruro están regulados por ácido
g-aminobutírico (GABA) y las anomalías en la unión y el metabolismo
de GABA pueden causar encefalopatías neonatales. Las
características distintivas de los trastornos del metabolismo del GABA incluyen la presencia de convulsiones al nacer, crecimiento
acelerado y GABA elevado en plasma y LCR. (6) La vigabatrina debe
considerarse como una opción terapéutica, pero puede ser beneficiosa
en algunos pacientes y perjudicial en otros. STXBP1 es uno de varios
genes implicados en la fusión de vesículas en las sinapsis y los
pacientes afectados pueden presentar encefalopatía y convulsiones.
Otros genes implicados en la función sináptica son TBC1D24 y SIK1.
CDKL5 y BRAT1 codifican proteínas involucradas en la señalización celular, que cuando se interrumpen dan como resultado un fenotipo epiléptico severo con letalidad temprana por BRAT1. Las encefalopatías epilépticas pueden definirse como epilepsia grave con disfunción neurológica permanente y suelen etiquetarse con un diagnóstico clínico como síndrome de Ohtahara, síndrome de West o síndrome de Dravet. Las características clínicas y los genes individuales asociados con las encefalopatías epilépticas se enumeran en Tabla 5. Pueden ocurrir como una característica principal de los trastornos de un solo gen y no necesariamente son causadas por convulsiones incontroladas. (7) (8) (9)
Aproximación investigacional
Cualquier buen enfoque para la
evaluación de una condición médica enfatiza la importancia de la
historia y un examen físico completo. El enfoque de las convulsiones
neonatales no es diferente. El médico evaluador debe comenzar por
obtener información sobre el episodio de la convulsión (inicio, tipo
de convulsión, factores desencadenantes, respuesta al tratamiento),
así como antecedentes familiares detallados y antecedentes de
nacimiento. Un examen físico completo debe incluir parámetros de
crecimiento (específicamente la circunferencia de la cabeza) y debe
prestarse atención al examen neurológico, examen de la piel y
presencia o ausencia de características dismórficas.
Se deben considerar las
evaluaciones de anomalías congénitas con ecocardiografía, ecografía
renal u otros estudios basados en la preocupación clínica. Los
estudios que se realizarán junto con una consulta de neurología
deben ser un EEG y una resonancia magnética cerebral (con
espectroscopia de resonancia magnética cuando sea posible). Si la
resonancia magnética es preocupante por malformaciones específicas,
la evaluación debe dirigirse a los trastornos genéticos asociados
con esas malformaciones.
El tipo de convulsión, otras características clínicas, la evaluación de laboratorio y los hallazgos de EEG o resonancia magnética cerebral deberían ayudar a reducir el diferencial y determinar el curso recomendado de las pruebas genéticas. Si se sospecha una afección genética específica, se deben solicitar pruebas específicas para ese trastorno. Un cariotipo puede detectar anomalías cromosómicas, como trisomías o grandes reordenamientos. Un microarray cromosómico evaluará las microdeleciones o duplicaciones. Si se sospecha un síndrome en particular, se pueden enviar pruebas para los genes asociados conocidos. Si se sospecha un IEM, se deben enviar evaluaciones bioquímicas (como se discutió anteriormente).
Si la anamnesis, la
exploración física, el electroencefalograma y la resonancia
magnética cerebral son inespecíficos, se debe considerar un
screening
amplio para etiologías metabólicas y genéticas de las convulsiones.
(10) A menudo se envía un panel de convulsiones multigénicas, que
evalúa docenas (a veces cientos) de genes asociados con las
convulsiones. Los paneles continúan aumentando de tamaño, a menudo
incluyen genes que aún pueden tener evidencia limitada de
correlación con las convulsiones. Los resultados deben analizarse en
el contexto del fenotipo del paciente, antes de asignar causalidad a
una mutación identificada. A menudo se considera la secuenciación
del exoma completo, que evalúa todos los exones (regiones
codificantes de proteínas) en todos los genes conocidos. La
secuenciación del genoma completo, como se denomina, evalúa el
genoma completo y está cada vez más disponible. En el entorno de la
unidad de cuidados intensivos neonatales , se puede considerar la
secuenciación urgente del exoma completo o del genoma completo,
porque la obtención de un diagnóstico puede afectar el manejo
inmediato.
Tenga en cuenta que el exoma
completo y la secuenciación del genoma completo pueden tener algunos
inconvenientes, incluida la falta de partes del genoma (lo que
conduce a resultados normales falsamente tranquilizadores) y el
descubrimiento de afecciones genéticas incidentales o no
relacionadas.
Opciones terapéuticas
Los avances en la
investigación traslacional han dado esperanzas para el desarrollo de
terapias dirigidas con genes específicos para los trastornos
convulsivos. Las terapias se centran en el control de las
convulsiones clínicas y subclínicas, así como en la mejora de los
resultados del neurodesarrollo a largo plazo. Muchos de los FAE que
se utilizan comúnmente ahora pueden tener efectos adversos en los
recién nacidos. Por ejemplo, con el uso de fenitoína o fenobarbital,
la preocupación preliminar es la apoptosis en los cerebros en
desarrollo. (11) Las terapias dirigidas son importantes, porque solo
los medicamentos que se sabe que son efectivos podrían usarse
preferentemente sobre los FAE inespecíficos comunes. Algunas
terapias dirigidas actualmente en uso se enumeran en la
Tabla 6.
Las epilepsias
dependientes de vitaminas se pueden tratar con suplementos
vitamínicos. Estos suelen ser resistentes a los FAE convencionales,
pero muestran una respuesta rápida a la administración de vitaminas.
La epilepsia dependiente de piridoxina puede tratarse con piridoxina
intravenosa u oral, pero esto puede resultar en depresión
respiratoria central en 20% de los pacientes. (12) Todo recién
nacido con convulsiones debe someterse a un ensayo de piridoxina
incluso si los trastornos metabólicos no son una etiología alta en
el diferencial. La deficiencia de piridoxina 59-fosfato oxidasa es
un trastorno similar que requiere tratamiento con la forma activa de
piridoxina, PLP. Las convulsiones que responden al ácido folínico,
como se denomina acertadamente, se resuelven con la suplementación
con ácido folínico.
Debido a la excitación anormal de los canales de potasio o sodio, las canalopatías son ejemplos de epilepsias con una etiología genética que pueden beneficiarse de terapia dirigida (canal específico). Se ha demostrado que la retigabina es beneficiosa para pacientes con alteraciones de la corriente de potasio en los canales KCNQ debido a mutaciones específicas en KCNQ2. La retigabina activa los canales de potasio, reduciendo la excitabilidad neuronal, disminuyendo así la actividad convulsiva. Se necesita precaución en el uso de este fármaco, porque puede tener un efecto perjudicial en pacientes con mutaciones de ganancia de función. (13) (14)
Un alto grado de sospecha (basado en los antecedentes familiares) y el inicio rápido de la terapia dirigida es fundamental para los bebés con ciertos trastornos convulsivos. Por ejemplo, en la deficiencia de cofactor de molibdeno, los outcomes tienen un amplio espectro según el momento de la intervención. Estos outcomes pueden variar desde "casi normales" si la terapia se inicia temprano, hasta una discapacidad severa del desarrollo si se inicia después de que ha ocurrido una lesión cerebral irreversible.
Conclusión
A medida que nuestro
conocimiento de la genética continúa progresando, aprendemos más
sobre la etiología de la epilepsia neonatal. La epilepsia neonatal
puede ser causada por malformaciones cerebrales estructurales, que
con frecuencia tienen una causa genética subyacente. Los IEM pueden
causar epilepsia neonatal debido a hipoglucemia, sustancias tóxicas
o malformaciones estructurales. Hay muchas causas de epilepsia de un
solo gen, incluidas las canalopatías y los genes implicados en el
metabolismo de los neurotransmisores. La epilepsia es una
característica común en muchos otros síndromes genéticos. A medida
que mejora nuestra capacidad para diagnosticar las causas genéticas
de las convulsiones, también lo hace nuestra capacidad para tratar
estas convulsiones mediante la terapia dirigida. Los recién nacidos
que presentan epilepsia sin factores perinatales evidentes que la
provoquen deben someterse a pruebas genéticas como parte de la
práctica clínica habitual.
Especificaciones de contenido neonatal-perinatal de Comité Estadounidense de Pediatría
Comprender el diagnóstico diferencial y la evaluación de las convulsiones neonatales.
Comprender el espectro de convulsiones en el recién nacido.
Tabla 1.- Syndromes with Structural Malformations of the Brain
|
Malformación congénita |
Sindrome asociado (gene) |
|
Corpus callosum agenesis/hypogenesis |
Aicardi syndrome (likely X-linked) |
| L1 syndrome (L1CAM) | |
|
Mowat-Wilson syndrome (ZEB2) |
|
|
Schinzel syndrome (KIF7) |
|
|
Tubulinopathies (TUBA1, TUBB2A, TUBB2B, TUBB3, TUBB, TUBG1) |
|
|
Type I lissencephaly (agyriapachygyria) |
Miller-Dieker syndrome (17p13.3 deletion) |
|
Isolated lissencephaly sequence (LIS1, TUBA1A, DCX) |
|
|
Type II lissencephaly |
X-linked lissencephaly with abnormal genitalia (ARX) |
|
Walker-Warburg syndrome (POMT1, POMT2, CRPPA, FKTN, FKRP, LARGE1) |
|
|
Fukuyama congenital muscular dystrophy (FKTN) |
|
|
Polymicrogyria (cortical dysplasia) |
1p36 deletion |
|
22q11.2 deletion |
|
|
Aicardi syndrome |
|
|
Bilateral frontoparietal polymicrogyria (ADGRG1) |
|
|
Goldberg-Shpritzen syndrome (KIAA1279) |
|
|
Joubert syndrome (many genes) |
|
|
mTORopathies (AKT3, CCND2, MTOR, PI4KA, PIK3CA, PIK3R2, PTEN) |
|
|
Tubulinopathies (TUBA1A, TUBB, TUBB2A, TUBB2B, TUBB3) |
|
|
Zellweger syndrome (multiple PEX genes) |
|
|
Schizencephaly |
Nonsyndromic schizencephaly (EMX2, SIX3, SHH, COL4A1) |
|
Megalencephaly |
Nonsyndromic megalencephaly (MTOR, SHH) |
|
Megalencephaly with gigantism (NSD1) |
|
|
Megalencephaly-capillary malformation syndrome, hemimegalencephaly (PIK3CA) |
|
|
Pretzel syndrome (LYK5/STRADA deletion) |
|
|
Microcephaly |
Microcephaly with pontine and cerebellar hypoplasia (CASK) |
|
Microcephaly, seizures, and developmental delay (PNKP) |
|
|
Holoprosencephaly |
Patau syndrome (trisomy 13) |
|
Nonsyndromic holoprosencephaly (SHH, ZIC2, SIX3, TGIF1) |
|
|
Septo-optic dysplasia |
Nonsyndromic septo-optic dysplasia (HESX1, OTX2, SOX2) |
|
Dandy-Walker malformation |
3q22-q24 deletion |
|
Aicardi syndrome |
|
|
Coffin-Siris syndrome (ARID1A, ARID1B, SMARCA4, SMARCB1, SMARCE1, and SOX11) |
|
|
Ciliopathies |
|
|
Edwards syndrome (trisomy 18) |
|
|
Fryns syndrome |
|
|
Patau syndrome (trisomy 13) |
|
|
Smith-Lemli-Opitz syndrome (DHCR7) |
|
|
Triploidy |
|
|
Walker-Warburg syndrome (POMT1, POMT2, CRPPA, FKTN, FKRP, LARGE1) |
Tabla 2.- Convulsiones de origen metabólico de inicio neonatal
| Tipo | Característica | Diagnóstico | Tratamiento |
|
Pyridoxine-dependent epilepsy |
Seizures refractory to common AEDs, hypothermia |
Low a-AASA and pipecolic acid |
Piridoxina |
|
Distonía |
ALDH7A1 gene testing |
|
|
|
PNPO deficiency |
|
Low CSF PLP |
PLP |
|
PNPO gene testing |
|
||
|
Nonketotic hyperglycinemia |
Prenatal hiccups, Lethargy, Hypotonia |
Elevated CSF-plasma glycine ratio (>0.08) |
Avoid valproic acid |
|
GLDC/AMT gene testing |
Sodium benzoate |
||
|
Dextromethorphan |
|||
|
Organic acidurias |
Hyperammonemia |
Urine organic acids, plasma amino acids, plasma acylcarnitines |
Low protein diet |
|
High anion gap metabolic acidosis |
Enzyme analysis |
Carnitina |
|
|
Molecular testing |
Cofactor supplementation |
||
|
Nitrogen scavengers |
|||
|
Urea cycle disorders |
Hyperammonemia |
Ammonia |
Low protein diet |
|
Plasma amino acids, urine orotic acid |
Nitrogen scavengers |
||
|
Enzyme analysis |
Citrulline/arginine |
||
|
Molecular testing |
|||
|
Peroxisomal disorders |
Structural brain malformations |
Very-long-chain fatty acids |
Standard AEDs |
|
Dysmorphic facies |
Phytanic acid |
||
|
Hypotonia |
RBC Plasmalogen |
||
|
Liver dysfunction |
Molecular testing |
||
|
Hearing loss |
|||
|
Folinic acid–responsive seizures |
Seizures refractory to common AEDs |
ALDH7A1/FOLR1 gene testing |
Folinic acid |
|
Transient response to pyridoxine |
|||
|
Biotinidase deficiency |
Alopecia, skin rash |
Biotinidase enzyme analysis |
Biotin |
|
Hearing and vision abnormalities |
BTD sequencing |
||
|
Hypotonia |
|||
|
Molybdenum cofactor deficiency |
Cerebral edema |
Urine sulphite |
Clinical trials for cPNP |
|
Developmental delays |
Urine sulfocysteine |
||
|
Sulphite oxidase deficiency |
Tone abnormalities |
Enzyme analysis |
Low protein diet(controversial) |
|
Progressive microcephaly |
Molecular testing |
||
|
Intellectual disability |
|||
|
Congenital disorders of glycosylation |
Hypotonia |
N- and O- Glycan analysis |
Symptomatic treatment |
|
Abnormal subcutaneous fat |
Molecular testing |
Mannose-1-phosphate trials |
|
|
Dysmorphic facies |
|
|
|
|
Structural brain malformations |
|
|
|
|
AASA - alfa - aminoadipic semialdehyde; AEDs : Antiepileptic drugs; CSF : cerebrospinal fluid; cPNP : cyclic pyranopterin; PLP : Pyridoxal phosphate ; PNPO : pyridoxine 59-phosphate oxidase deficiency; RBC : red blood cell. |
|||
Tabla 3.- Características MRI / MRS de los trastornos metabólicos
|
METABOLIC DISORDER |
MRI FINDING |
|
Disorders of creatine metabolism |
Decreased/absent creatine |
|
|
± increased GAMT |
|
GABA transaminase deficiency |
Elevated GABA in basal ganglia |
|
Glutaric acidemia type 1 |
Frontotemporal atrophy, widened “Batwing” sylvian fissures |
|
Maple syrup urine disease |
Localized edema in cerebellar white matter, dorsal brain stem, cerebral peduncles, posterior limb of internal capsule |
|
Mitochondrial disorders, congenital lactic acidosis |
Elevated lactate peak |
|
Peroxisomal disorders |
Decrease in white matter volume, decrease of myelination, ventricular enlargement, abnormal gyration |
|
Nonketotic hyperglycinemia |
Defects of the corpus callosum, elevated glycine peak |
|
Organic acidemias |
Elevated glycine and/or lactate peaks ; Cerebral edema |
|
Molybdenum cofactor deficiency |
Severe cerebral edema, acute infarction of the globus pallidi and subthalamic regions |
|
GABA = g-aminobutyric acid; GABT = guanidinoacetate peak; MRI = magnetic resonance imaging; MRS = magnetic resonance spectrography. |
|
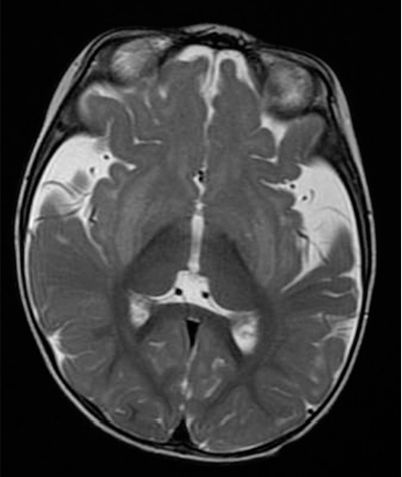
Figura.- Resonancia magnética (T2 axial) que muestra atrofia
frontotemporal y fisuras de Silvio ensanchadas en un paciente con
acidemia glutárica tipo 1.
Tabla 4.- Sindromes genéticos asociados con Epilepsia neonatal
|
TYPE OF CONDITION |
SYNDROME |
GENETIC ETIOLOGY/GENE |
OTHER ASSOCIATED FEATURES (IN THE NEONATAL PERIOD) |
|
Chromosomal |
Down syndrome |
Trisomy 21 |
Hypotonia , Congenital heart disease; Dysmorphic features |
|
Patau syndrome |
Trisomy 13 |
Cleft lip/palate; Omphalocele ; Holoprosencephaly; Congenital heart disease; Dysmorphic features |
|
|
Edwards syndrome |
Trisomy 18 |
Clenched hands, overlapping fingers ; Rockerbottom feat; Congenital anomalies; Dysmorphic features |
|
|
22q11.2 deletion syndrome |
Deletion of 22q11.2 |
Congenital heart defects; Palate abnormalities; Hypocalcemia; Immune deficiency |
|
|
Wolf-Hirschhorn syndrome |
Deletion of 4p16.3 |
Poor growth ; Hypotonia; Dysmorphic features |
|
|
Neurocutaneous |
Tuberous sclerosis |
TSC1, TSC2 |
Skin abnormalities: Hypopigmented macules, shagreen patches, angiofibromas Hamartomas in the brain, heart, kidneys, retina |
|
Sturge-Weber |
GNAQ |
Port wine stain Glaucoma Leptomeningeal angioma |
|
|
Incontinentia pigmenti |
IKBKG |
Blistering rash in neonatal period (evolves eventually into linear hypopigmentation) |
|
|
Hypomelanosis of Ito |
Mosaicism for aneuploidy or other chromosomal anomalies |
Hypopigmented whirls along lines of Blaschko; Other congenital anomalies |
|
|
Otras |
COL4A1-related |
COL4A1 |
Small vessel disease; Porencephaly; Eye defects. |
|
Pitt-Hopkins |
TCF4 |
Episodic hyperventilation; Feeding issues, constipation; Eye abnormalities; Microcephaly. |
|
|
Coffin-Siris |
ARID1A, ARID1B, SMARCA4, SMARCB1, SMARCE1, and SOX11 |
Hypoplasia of 5th digit; Dysmorphic features; Brain malformations; Poor growth; Hypotonia |
|
|
Aicardi-Goutieres |
ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, or TREX1, IFIH1 |
Early-onset encephalopathy; Glaucoma; Liver dysfunction |
Tabla 5.- Encefalopatías epilépticas neonatales
|
|
CLINICAL CHARACTERISTICS |
ASSOCIATED GENES |
|
Ohtahara syndrome |
Intractable seizures ; Developmental delay ; EEG: burst suppression |
STXBP1;ARX; SLC25A22; KCQN2; SCN2A |
|
Early myoclonic encephalopathy |
Erratic myoclonus, refractory partial seizures ; Psychomotor developmental delay, hypotonia, peripheral; neuropathy; EEG: burst suppression |
SLC25A22 ; MOCS1; SEPSECS |
|
Epilepsy of infancy with migrating focal seizures |
Refractory focal seizures ; Severe psychomotor delay; EEG: frequent slow waves shifting from one hemisphere to another |
KCNT1; SCN1A; SCN2A; SLC25A22 |
|
West syndrome and Dravet syndrome usually have an age at onset of more than 1 month. (9) EEG¼electroencephalography. |
||
Tabla 6.- Seizure Disorders with Targeted Therapies
|
DISORDER |
TREATMENT |
|
Pyridoxine-dependent epilepsy (ALDH7A1) |
Vitamin B6 |
|
DEPDC5-related epilepsy |
mTOR inhibitors (sirolimus, everolimus) |
|
Cerebral folate transport deficiency (FOLR1) |
Folinic acid |
|
Guanidinoacetate methyltransferase deficiency (GAMT) |
Creatine, ornithine |
|
Early-onset epileptic encephalopathy (KCNQ2) |
Retigabine, carbamezapine |
|
KCNT1-related epilepsy |
Quinidine |
|
Molybdenum cofactor deficiency (MOCS1) |
Clinical trial for cyclic pyranopterin |
|
SCN1A-related epilepsy |
Avoid sodium channel blockers |
|
Early infantile epileptic encephalopathy 11 (SCN2A) |
Phenytoin, sodium channel blockers |
|
Tuberous sclerosis (TSC1, TSC2) |
Vigabatrin, mTOR inhibitors (sirolimus, everolimus) |
|
mTOR = mammalian target of rapamycin. |
|
Referencias
-
Shellhaas RA, Wusthoff CJ, Tsuchida TN, et al; Neonatal Seizure Registry. Profile of neonatal epilepsies: characteristics of a prospective US cohort. Neurology. 2017;89(9): 893–899
-
Pisani F, Percesepe A, Spagnoli C. Genetic diagnosis in neonatalonset epilepsies: back to the future. Eur J Paediatr Neurol. 2018;22(3):354–357
-
Margari L, Palumbi R, Campa MG, et al. Clinical manifestations in children and adolescents with corpus callosum abnormalities. J Neurol. 2016;263(10):1939–1945
-
Wolf NI, Bast T, Surtees R. Epilepsy in inborn errors of metabolism. Epileptic Disord. 2005;7(2):67–81
-
Gospe SM Jr. Neonatal vitamin-responsive epileptic encephalopathies. Chang Gung Med J. 2010;33(1):1–12
-
Koenig MK, Hodgeman R, Riviello JJ, et al. Phenotype of GABA-transaminase deficiency. Neurology. 2017;88(20): 1919–1924
-
Sands TT, McDonough TL. Recent advances in neonatal seizures. Curr Neurol Neurosci Rep. 2016;16(10):92
-
Liu J, Tong L, Song S, et al. Novel and de novo mutations in pediatric refractory epilepsy. Mol Brain. 2018;11(1):48
-
Gürsoy S, Erçal D. Diagnostic approach to genetic causes of earlyonset epileptic encephalopathy. J Child Neurol. 2016;31(4): 523–532
-
Axeen EJT, Olson HE. Neonatal epilepsy genetics. Semin Fetal Neonatal Med. 2018;23(3):197–203
-
Forcelli PA, Janssen MJ, Vicini S, Gale K. Neonatal exposure to antiepileptic drugs disrupts striatal synaptic development. Ann Neurol. 2012;72(3):363–372
-
Surtees R, Wolf N. Treatable neonatal epilepsy. Arch Dis Child. 2007;92(8):659–661
-
Milh M, Cacciagli P, Ravix C, et al. Severe neonatal seizures: from molecular diagnosis to precision therapy? Rev Neurol (Paris). 2016;172(3):171–173 14.
-
Mulkey SB, Ben-Zeev B, Nicolai J, et al. Neonatal nonepileptic myoclonus is a prominent clinical feature of KCNQ2 gain-offunction variants R201C and R201H. Epilepsia. 2017;58(3):436–445