
Sexo ambiguo
Marzo 2016
Ambiguous Genitalia : Evalución y manejo en el recién nacido
Bonnie McCann-Crosby, NeoReviews Marzo 2016
Introducción
Los neonatos cuyos genitales no pueden ser
clasificados claramente como fenotipo masculino o femenino se considera que
tienen un trastorno del desarrollo sexual (TDS). La evaluación de un recién
nacido con genitales ambiguos puede ser un desafío y requiere pronta investigación para determinar la causa subyacente. Se recomienda un enfoque con
equipo multidisciplinario que incluye neonatologo, endocrinologo, ginecologo, urologo. genética,
ética, trabajadora social y psicóloga cuando se evalúan
recién nacidos con genitales ambiguos. Los padres siempre deben ser incluídos en
las discusiones sobre la evaluación y manejo y deben ser actores clavse en la
decisión de asignación de sexo.
Los TDS se pueden clasificar en 3 grandes categorías : TDS 46, XX ;
TDS 46, XY y TDS cromosoma sexual. Un enfoque sistémico en
la evaluación de los genitales ambiguos puede ayudar a descubrir la etiología
subyacente en muchos casos pero algunas etiologías pueden no ser identificadas.
Este artículo se centra en el proceso de desarrollo sexual normal y describe la
evaluación clínica y el tratamiento de recién nacidos con genitales ambiguos.
Diferenciación sexual normal
Durante la embriogésis temprana, las gónadas y las estructuras internas de los fetos masculinos y femeninos son fenotípicamente idénticos y bipotenciales.
La gónada bipotencial se diferencia en ya sea un ovario o un testíulo, en función de la expresión génica específica. El primer paso en el desarrollo sexual masculino es expresión de la región determinante del sexo en el cromosoma Y (SRY), que se produce alrededor de la sexta semana de gestación. SRY desencadena genes determinantes del sexo adicionales y factores de transcripción que son necesarias para la maduración y la diferenciación de las células tipos en el testículo, incluyendo las células de Sertoli y de Leydig.
Alrededor de la octava semana de
gestación, los testículos primitivos comienzan a producir hormonas. La
producción de testosterona por las células de Leydig conduce al desarrollo de
los conductos de Wolff en los genitales masculinos internos (epidídimo,
conductos deferentes y títulos seminíferos). La producción de la hormona anti-Mulleriana
(AMH) por las células de Sertoli conduce a la involución de los conductos
internos de Muller (femeninos) . El desarrollo de los genitales externos
masculinos es dependiente de la actividad de la dihidrotestosterona (DHT), que
se convierte a partir de la testosterona por la enzima 5 alfa - reductasa. DHT
induce el desarrollo del pene desde el tubérculo genital, desarrollo de la
uretra desde el seno urogenital y la fusión de los pliegues labioescrotales para
formar el escroto.
En ausencia de SRY, las células gonadales en el embrión XX se transforman en
células de la granulosa y la teca del ovario. La ausencia de AMH (hormona
antimulleriana) permite que los conductos de Muller se conviertan en los
genitales femeninos internos (útero, trompas de Falopio y tercio superior
de la vagina) y hay regresión de los conductos de Wolff . Sin testosterona ni
DHT, el tubérculo genital formará el clítoris, los pliegues uretrales se
convierten en los labios menores, y los pliegues labioescrotales se convierten
en los labios mayores, lo que causa el fenotipo femenino externo.
TDS 46 , XX
El TDS 46, XX puede subdividirse en desórdenes por exceso de andrógenos o desórdenes del desarrollo ovárico. Estas condiciones se resumen en Tabla 1. La causa más común de ambigedad genital en una 46, XX femenina es la hiperplasia suprarrenal congénita (CAH). CAH es un trastorno autosómico recesivo con un defecto en la síntesis de cortisol.
Tabla 1.- TDS 46, XX que puede presentarse con genitales ambiguos
La causa más común de hiperplasia
suprarrenal congénita es la deficiencia de 21-hidroxilasa, que representa el 95%
de los casos; Sin embargo, la deficiencia de 11- hidroxilasa y la deficiencia
3b-hidroxiesteroide deshidrogenasa (HSD) también pueden causar virilización de
una RN de sexo femenino. El bloqueo en la producción de cortisol causa desvío de
los precursores de cortisol hacia la vía de andrógenos, lo que causa
virilización de los genitales externos. Las niñas 46, XX
con hiperplasia suprarrenal congénita tienen el desarrollo genital interno
femenino normal.
Las niñas que nacen con CAH pueden presentar una amplia gama de ambiguedad genital, desde leve clitoromegalia hasta apariencia fenotípica masculina con sacos escrotales vacíos. La Figura 1 muestra una RN femenina severamente virilizada con deficiencia de 21-hidroxilasa.
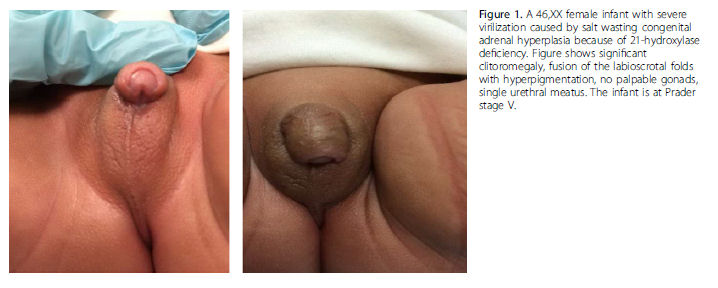
Los RN con deficiencia de 21 hidroxilasa clásica también tienen producción deficiente de aldosterona y se pueden presentar con una crisis perdedora de sal (hiponatremia, hipopotasemia, hipoglucemia, hipovolemia, shock). Todos los Estados tienen actualmente programas de screening neonatal que miden los niveles de 17-hidroxiprogesterona, que se encuentran elevados en casos de deficiencia de 21-hidroxilasa.
La deficiencia de aromatasa es una condición en la cual los pacientes son incapaces de convertir precursores de andrógenos a estrógenos. La deficiencia de aromatasa en la placenta causa virilizacion de la madre y del feto. Las causas maternas de exceso de andrógenos que pueden causar virilización de una niña 46, XX incluyen la ingestión de andrógenos, progestinas, tumores suprarrenales virilizantes , tumores de ovario o luteomas.
Trastornos del desarrollo de ovario en una niña 46, XX que se puede presentar con genitales ambiguos incluyen TDS ovotesticular y TDS 46, XX testicular. En el TDS ovotesticular la gónada contiene tanto tejido ovárico como testicular. Los pacientes con TDS ovotesticular pueden presentar un espectro de ambiguedad genital, así como tambien ambas estructuras de conductos internos masculinos y femeninos. En el TDS 46, XX testicular, ambas gónadas se desarrollan en testículos y estos pacientes no tienen ovarios ni componentes mullerianos. Estos pacientes pueden presentar anomalías genitales leves incluyendo hipospadias y testículos no descendidos, aunque en muchos casos son fenotípicamente varones normales que presentan más adelante en la vida infertilidad.
TDS 46, XY
TDS 46, XY se puede clasificar en trastornos del desarrollo testicular y trastornos de la síntesis o acción de los andrógenos. Estas condiciones se resumen en la Tabla 2.
Tabla 2.- TDS 46, XY que pueden presentarse con genitales ambiguos

Los trastornos del desarrollo testicular
incluyen disgenesia gonadal, regresión gonadal o sindrome de desaparición de
los testículos y TDS XY ovotesticular. En la disgenesia gonadal 46,
XY completa
(sindrome de Swyer) no se produce desarrollo testicular y los pacientes
se muestran como hembras fenotípicas con gónadas bilaterales y estructuras de Muller normales.
Los pacientes con 46, XY disgenesia
gonadal parcial tienen algún grado de desarrollo testicular y pueden presentar
fenotipos internos y externos variables dependiendo de cuan funcionales son los testículos. Varios sindromes, incluyendo displasia camptomelica, sindrome de
Frasier y sindrome de Denys-Drash, están asociados con disgenesia gonadal XY.
El sindrome de desaparición de testículos o sindrome de regresión testicular es una condición en la que los testículos están ausentes en un individuo 46, XY. Se cree que estos individuos tienen desaparición o regresión de los testículos in útero después de un insulto tal como torsión o trombosis vascular. La apariencia de los genitales externos depende del momento de la regresión testicular en relación con el desarrollo sexual : la regresión testicular antes de las 8 semanas de gestación causa una mujer fenotípica, la pérdida entre 8 y 10 semanas causa genitales ambiguos y la pérdida después de 12 semanas causará genitales externos masculinos normales.
TDS XY ovotesticular es una condición en la cual un individuo XY tiene tanto tejido
ovárico como testicular. Los pacientes con TDS XY ovotesticular con mayor
frecuencia se presentan con genitales ambiguos o hipospadias severas.
Los trastornos de la acción de andrógenos
incluyen la insensibilidad completa o parcial a andrógenos. Estas condiciones
son el resultado de mutaciones en el gen del receptor de andrógenos. El fenotipo
de los pacientes con insensibilidad a andrógenos depende del grado de respuesta tisular a la actividad de andrógenos. Los pacientes
con completa insensibilidad a andrógenos tienen genitales externos femeninos
con una bolsa vaginal ciega. Estos individuos se presentan típicamente en la
adolescencia con amenorrea primaria, aunque en algunos casos se identifican
antes debido a aumentos de volumen inguinales o labiales que contienen los testículos. Los pacientes con insensibilidad parcial a
andrógenos pueden presentar una amplia gama de fenotipos, desde genitales
ambiguos a fenotipo masculino que se presentan con infertilidad en la edad
adulta.
Los trastornos de síntesis de andrógenos incluyen defectos enzimáticos de biosíntesis de testosterona, aplasia o hipoplasia de células de Leydig y deficiencia de 5a-reductasa. Defectos enzimáticos en la glándula suprarrenal o testículos pueden causar disminución de la producción de testosterona y causan subvirilización en individuos 46 , XY. La vía de biosíntesis adrenal se muestra en Figura 2. Como se muestra en la figura, la síntesis de la testosterona comienza con el colesterol. La deficiencia de 7- dehidrocolesterol reductasa causa síntesis alterada de colesterol y causa el sindrome de Smith-Lemli-Opitz. Este sindrome se caracteriza por manifestaciones clínicas variables incluyendo defectos cardíacos, paladar hendido, sindactilia, polidactilia y ambiguedad genital.
Figura 2.- Vías de biosíntesis adrenal.
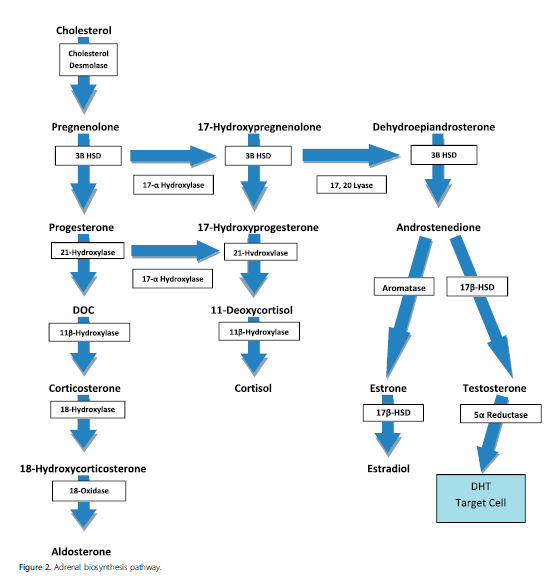
La conversión de colesterol a pregnenolona depende de la proteína de regulación aguda de esteroidogénesis
(StAR) y P450 scc; por lo tanto, los defectos en estas 2 enzimas pueden
causar hombres sub virilizados. Estos pacientes suelen aparecer como
hembras fenoticas con testículos no descendidos. Los individuos con mutaciones
StAR tienen insuficiencia suprarrenal y acumulación de lípidos y
agrandamiento de sus glándulas suprarrenales. Los pacientes con P450 scc tienen un
fenotipo similar al de los individuos con deficiencia de StAR. 3
Beta - HSD tipo II es la enzima que convierte dehydroepiandrostenediona a androstenediona.
Las deficiencias en 3 Beta - HSD tipo II pueden
causar hipospadias, micropene, subvirilización / ambiguedad más grave , además
de deficiencia de cortisol y de mineralocorticoides. Las mutaciones en el gen CYP17
pueden causar deficiencia en las actividades de 17a-hidroxilasa y 17,20
liasa. Los individuos XY con deficiencia de 17 alfa hidroxilasa / 17 ,20 liasa se pueden
presentar con diferentes grados de subvirilización que van desde micropene,
hipospadias perineal y criptorquidia a un fenotipo femenino externo completo.
La deficiencia de
17 Beta - HSD de tipo III causa incapacidad para convertir androstenodiona en testosterona. Estos individuos XY pueden
presentarse como
mujer o tener fenotipo externo ambiguo.
Las mutaciones en el receptor de hormona luteinizante (LH) / gonadotropina coriónica humana (hCG) pueden causar
aplasia o hipoplasia de células de Leydig testiculares. Sin la producción normal
de testosterona por las células de Leydig, se producirá subvirilización del varón XY. El grado de subvirilización depende del grado de la hipoplasia de
células de Leydig.
La deficiencia de 5a-reductasa causa un defecto en la conversión de testosterona a DHT. Sin los niveles adecuados de DHT para virilizar los genitales externos, los individuos XY pueden presentar genitales ambiguos, micropene, hipospadias o genitales externos femeninos. Estos pacientes pueden tener virilización espontánea durante la pubertad debido al aumento de los niveles de testosterona.
TDS cromosoma sexual
El TDS de Cromosoma sexual incluye el sindrome de Turner (45, X), sindrome de Klinefelter (47, XXY) y cariotipos de mosaico (por ejemplo, 45, X / 46, XY ; 46, XX / 46, XY). Los pacientes con sindrome de Turner y Klinefelter no mosaico no se presentan con genitales ambiguos, mientras que aquellos con los cariotipos de mosaico, tales como 45, X / 46, XY pueden presentarse con diferentes grados de ambiguedad genital en función del grado de desarrollo y funcional de sus testículos.
Evaluación diagnóstica
Las características clínicas que originan la sospecha de un TDS incluyen genitales femeninos aparentes con clitoromegalia, masa inguinal, masa labial o fusión labial posterior; micropene con testículos no palpables bilaterales, hipospadias con testículos no descendidos, penoescrotal aislado o hipospadias perineoescrotal; o una discordancia entre el cariotipo prenatal y la apariencia genital.
Clitoromegalia se define como longitud del clítoris mayor de 9 mm , ancho del clítoris mayor de 6 mm en un RN a término. Una longitud del pene estirado que esté bajo 2,5 SD por debajo de la media para la edad se considera que es un micropene. La evaluación de un recién nacido con genitales ambiguos requiere una historia clínica cuidadosa , examen físico, pruebas de laboratorio y estudios de imagen.
Debe preguntarse a los padres sobre consanguinidad, la que puede indicar un
mayor riesgo de trastornos autosómicos recesivos tales como hiperplasia
suprarrenal congénita. Una historia de muertes infantiles inexplicadas en la
familia tambien puede ser preocupante para diagnosticar CAH con pérdida de sal.
Tambien se debe obtener una historia familiar de genitales ambiguos, anomalías
urológicas , infertilidad / amenorrea femenina . Las madres deben ser
preguntadas por el uso de cualquier medicamento , exposición a factores
ambientales que podrían causar virilización de un neonato de sexo
femenino. Debe observarse virilización materna, ya que podría sugerir un
tumor materno que produce exceso de andrógenos.
Se deben obtener los resultados de las pruebas prenatales,
incluyendo la ecografía y determinación del cariotipo. El
examen físico debe comenzar con una evaluación general. Como se mencionó
anteriormente, algunos sindromes se asocian con la ambiguedad genital y
tienen rasgos físicos característicos. Cualquier rasgo o malformación dismórfica debe tenerse en cuenta, ya que pueden sugerir un trastorno
genético subyacente. Los defectos de la línea media se pueden asociar con anomalías
hipotálamo-pituitaria y pueden causar hipogonadismo hipogonadotrófico. La
hipertensión y la deshidratación pueden estar asociadas con algunas formas de
hiperplasia suprarrenal congénita, por lo que es importante evaluar el estado
de hidratación y la presión arterial.
La ictericia puede ser un signo de deficiencia de cortisol o hipopituitarismo. El examen de los genitales externos debe incluir el desarrollo del tubérculo genital, que forma el pene en los varones y el clítoris en las mujeres. Debe observarse el grado de fusión de los pliegues labioescrotales así como tambien la textura y la pigmentación de la piel de los genitales. Es importante palpar los pliegues labioescrotales y regiones inguinales bilateralmente en búsqueda de presencia de gónadas. Una gónada palpable externa sería o un testículo o un ovotestis. El tamaño y la ubicación de las gónadas se deben documentar. La longitud y anchura del falo estirado deben ser medidos y la presencia o ausencia de curvatura peneana deben tenerse en cuenta.
La ubicación de la abertura de la uretra o del seno urogenital debe ser determinada
(¿ hay aberturas vaginales y uretrales separadas o hay un canal común). El grado
de hipospadias se debe documentar si está presente (está la abertura uretral proximal / perineal,
en la mitad o distal / glandular en la
superficie ventral de la estructura fálica ?). En contraste con hipospadias,
las epispadias son la apertura de la uretra o del seno urogenital en la superficie
dorsal de la estructura fálica. Las epispadias son una ocurrencia rara y pueden ser
parte de un espectro de condiciones en las que existe falta de fusi? de
?ganos abdominales o p?vicos, incluyendo los genitales externos.
Todos los pacientes con genitales ambiguos o con pregunta de asignación de sexo requieren un análisis cromosómico de rutina como la investigación inicial. Debe ser obtenido dentro de las primeras 24 horas después del nacimiento y el laboratorio debe ser notificado para acelerar los resultados.
Se recomienda un análisis de microarrays cromosómico para buscar pequeñas deleciones o duplicaciones que pueden no ser identificadas en el cariotipo de rutina. Para los pacientes con genitales ambiguos y gónadas no palpables debe haber una alta sospecha de hiperplasia suprarrenal congénita. La evaluación inicial debe incluir la medición de electrolitos séricos en busca de pérdida de sal, glucosa sérica que puede ser baja en los casos de deficiencia de cortisol y 17-hidroxiprogesterona para evaluar la deficiencia de 21- hidroxilasa.
Puede ser necesario un test de
estimulación de corticotropina (ACTH) para evaluar la respuesta de cortisol y deficiencias enzimáticas
que pueden causar CAH. Se puede hacer mediante la obtención de la línea de base y
los niveles de cortisol estimulados por ACTH , 17-hidroxiprogesterona, 17-hidroxipregnenolona,
progesterona, androstenediona, dehidroepiandrosterona, desoxicorticosterona,
11-desoxicortisol y testosterona. El test de estimulación con ACTH no debe
retrasar el tratamiento en casos de sospecha de hiperplasia suprarrenal
congénita. En cualquier caso sospechoso de hiperplasia suprarrenal congénita,
los niveles de renina y aldosterona tambien deben obtenerse. Las pruebas
adicionales incluyen gonadotropinas basales (LH y hormona folículo
estimulante), que pueden ser bajas en los casos de hipogonadismo hipogonadotrófico e indicativas de posible deficiencia pituitaria.
El hipogonadismo hipogonadotrópico puede ser
una causa de micropene en individuos 46, XY. Un nivel de AMH es un marcador
fiable de la presencia y función del tejido testicular y puede ser útil en la
evaluación de los individuos XY subvirilizados. Los niveles de AMH serán bajos en
los casos de testículos desaparecidos, disgenesia XY gonadal o sindrome de
conducto de Muller persistente. Los niveles de AMH pueden ser elevados en los
casos de insensibilidad a los andrógenos e hipogonadismo hipogonadotrófico. Se
utiliza un test de hCG-estimulación para evaluar la producción de testosterona
a partir de tejido testicular. El test de hCG-estimulación requiere medición
basal y niveles estimulados de testosterona y DHT. La relación
de testosterona a DHT será elevada en los casos de deficiencia de 5a-reductasa.
Una respuesta inadecuada a la estimulación con hCG en un individuo XY puede ser indicativo de disgenesia gonadal, TDS ovotesticular , defecto del receptor de LH o hipogonadismo hipogonadotrófico. Los altos niveles de testosterona en un paciente 46, XY subvirilizado deben plantear la sospecha de insensibilidad a andrógenos y deben considerarse pruebas genéticas. La anatomía interna de cualquier paciente con genitales ambiguos debe ser evaluada con ultrasonografía abdominal / pélvica o resonancia magnética. La imagenología puede identificar la presencia o ausencia de estructuras de Muller (útero) y la localización del tejido gonadal. Puede ser necesaria una biopsia gonadal biopsia para determinar el tipo de tejido gonadal en casos de sospecha de TDS ovotesticular o disgenesia gonadal.
Manejo
La incertidumbre de género o sexo de un neonato es estresante y molesta para las familias. La implicación inmediata de un equipo multidisciplinario que tenga conocimiento en TDS es esencial en la evaluación de estos pacientes. Este equipo debe incluir endocrinologo, urologo, ginecologo, genética, psicologo, trabajo social y ética.
Descartar las emergencias que amenazan la vida, tales como CAH debe ser la prioridad inicial. Los padres deben participar en todas las discusiones relativas a la asignación de sexo y deben estar actualizados sobre los resultados de pruebas a medida que estén disponibles. Aunque los padres están dispuestos a decirle a sus amigos y familiares el sexo de su recién nacido, es importante que la consideración cuidadosa y exhaustiva evaluación estén completas antes de tomar una decisión de asignación de sexo.
Antes de realizar la asignación de sexo final, es importante utilizar términos neutros de género o sexo cuando se habla del neonato, tales como "bebé "; en lugar de utilizar "ella" o "él ." Se recomienda que los padres se reúnan con un psicólogo que pueda ayudarles con la superación de las pruebas y les proporcione orientación sobre la manera de hablar con amigos y familiares acerca de su recién nacido. Muchos factores, incluyendo el sexo fenotípico (apariencia de los genitales externos e internos), el sexo genotítipico (cariotipo), el sexo hormonal (perfil hormonal: testosterona, DHT, perfil de esteroides suprarrenales), sexo reproductivo (potencial de tener hijos biológicos) y la percepción de los padres , influyen en la asignación sexual. Discutir cada uno de estos componentes del desarrollo sexual con la familia puede ayudar a desmitificar el proceso y reducir la ansiedad para las familias a medida que participan en el proceso de asignación de sexo.
La intervención quirúrgica (gonadoplastía, vaginoplastía , gonadectomía) deben retrasarse hasta que se establezca un diagnóstico claro y debe ser realizado solamente por un cirujano experimentado.
Los beneficios de cualquier procedimiento quirúrgico o médico deben superar
ampliamente los riesgos y todos los procedimientos innecesarios deben ser
retrasados hasta que el niño tenga edad suficiente para tomar una decisión
informada.
na vez que se toma una decisión de asignación de sexo, el niño y su familia
necesitarán seguimiento a largo plazo con médicos que tengan
experiencia con TDS. La educación continua es esencial ya que muchos de estos
pacientes requerirán cirugía y terapia hormonal. Es importante evaluar la satisfacción del paciente y de los padres con la decisión de asignación de sexo. Se
recomienda evaluación psicológica a intervalos regulares para detectar
problemas de salud mental tales como disforia de género y para proporcionar
apoyo a las familias.
Conclusiones
El hallazgo de genitales ambiguos en el recién nacido puede ser angustiante tanto para los clínicos tratantes como para las familias. Un enfoque paso a paso completo para el diagnóstico que involucra a un equipo multidisciplinario es necesario. Cada caso de ambiguedad genital es único y una comprensión básica del diagnóstico diferencial de TDS puede ayudar a guiar la evaluación de estos recién nacidos. La comunicación abierta entre el equipo médico y las familias es importante y las familias deben participar en todas las decisiones sobre asignación de sexo y tratamiento quirúrgico.
Referencias
Ahmed SF, Rodie M. Investigation and initial management of ambiguous genitalia. Best Pract Res Clin Endocrinol Metab. 2010;24 (2):197?218
Axelrad ME, Berg JS, Coker LA, et al. The gender medicine team: ?it takes a village.? Adv Pediatr. 2009;56:145?164
Barbaro M, Wedell A, Nordenstr? A. Disorders of sex development. Semin Fetal Neonatal Med. 2011;16(2):119?127
Lambert SM, Vilain EJ, Kolon TF. A practical approach to ambiguous genitalia in the newborn period. Urol Clin North Am. 2010;37 (2):195?205
Lee PA, Houk CP, Ahmed SF, Hughes IA; International Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. Consensus statement on management of intersex disorders. Pediatrics. 2006;118(2): e488?e500
MacLaughlin DT, Donahoe PK. Sex determination and differentiation. N Engl J Med. 2004;350(4):367?378
Mieszczak J, Houk CP, Lee PA. Assignment of the sex of rearing in the neonate with a disorder of sex development. Curr Opin Pediatr. 2009;21(4):541?547
Murphy C, Allen L, Jamieson MA. Ambiguous genitalia in the newborn: an overview and teaching tool. J Pediatr Adolesc Gynecol. 2011;24 (5):236?250
Parent Resources from the AAP at HealthyChildren.org