Infecciones Perinatales
Septiembre 2021
Infecciones en la interfaz materno-fetal: descripción general de patogenia y defensa
Christina J. Megli Division of Maternal–Fetal Medicine, University of Pittsburgh USA Nature Reviews | Microbiology Agosto 2021
Las infecciones durante el embarazo pueden tener consecuencias devastadoras para la madre embarazada y el feto en desarrollo. La Transmisión vertical, definida como infección del feto desde el huésped materno es una de las principales causas de morbilidad y mortalidad durante el embarazo. En algunos casos, las infecciones bacterianas, virales y parasitarias inducen outcomes nefastos en el feto. Las secuelas de las infecciones en el embarazo incluyen efectos teratogénicos, que provocan anomalías congénitas; restricción del crecimiento, muerte fetal, aborto espontáneo y muerte neonatal; prematuridad y morbilidad materna (Figura 1).
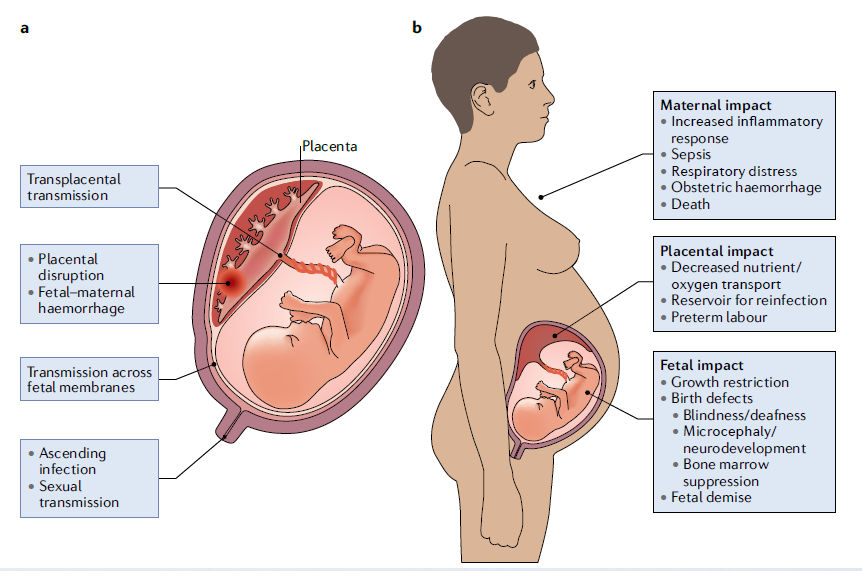
Figura 1.- Rutas de transmisión a través de la placenta y consecuencias de la infección.
a.- Los patógenos TORCH (Toxoplasma gondii, otros, virus de la rubéola, citomegalovirus, virus del herpes simple) pueden acceder al compartimento intraamniótico a través de múltiples mecanismos, incluyendo transmisión transplacentaria directa, daño o rotura placentaria y / o hemorragia fetal-materna. Además, los patógenos se pueden transmitir ascendiendo por el tracto genital.
b.- Las infecciones durante el embarazo pueden afectar al huésped materno, al feto y / o a la propia placenta. Los resultados de la infección y la respuesta inflamatoria tienen consecuencias en cada sitio.
La transmisión vertical de patógenos a
través de la interfaz materno-fetal puede causar infección fetal, la que puede
alterar la organogénesis y se asocia con anomalías congénitas de todos los
sistemas de órganos principales (tabla
1).
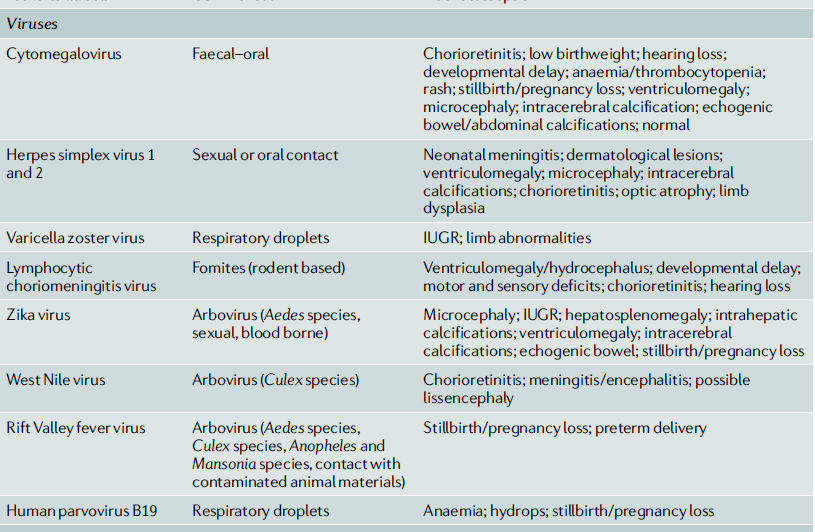
Tabla 1.- Patógenos TorcH y características de sus infecciones


Las anomalías congénitas están presentes en ~ 3% de los nacidos vivos y la proporción atribuible a infección no está bien definida1–3.
La pérdida del embarazo por aborto espontáneo o muerte fetal (definida como muerte fetal intrauterina después de 20 semanas) también puede ser causada por una infección. Aproximadamente el 10-30% de todos los mortinatos tienen etiología infecciosa4-7, aunque las bajas tasas de pruebas diagnósticas para infecciones durante el embarazo pueden resultar en una subestimación de este valor. La tasa de pérdida del embarazo varía con la edad gestacional en el momento de la infección y según el patógeno específico. Por ejemplo, la infección por Treponema pallidum provoca pérdida del embarazo o muerte fetal hasta en el 50% de los casos8, mientras que la infección por parvovirus B19 provoca la pérdida del embarazo o la muerte fetal en < 3% 9. Los mecanismos de pérdida del embarazo en el contexto de infecciones por "TORCH" (Toxoplasma gondii, otro, virus de la rubéola, citomegalovirus, virus del herpes simple) pueden ser mediados por patógenos, mediadas por placenta y / o pueden ser a través de un parto previable inducido por inflamación.
También hay resultados de infecciones
congénitas que no se manifiestan hasta después del parto. Estos pueden incluir
pérdida de audición, retrasos en el desarrollo y / o ceguera, como se detalla a
continuación. La inflamación iniciada por una infección del huésped materno es
también una causa conocida de trabajo de parto prematuro y puede resultar en un
parto previo o secuelas de prematuridad10 con consecuencias de por vida para el
neonato. Los recién nacidos pueden tener secuelas de infecciones maternas
incluso con desarrollo a término11, lo que demuestra que la respuesta fetal a la
infección no se limita a los efectos de la prematuridad.
La sepsis de inicio temprano, causa importante de morbilidad y mortalidad
neonatal, particularmente en los recién nacidos prematuros o de bajo peso al
nacer, está fuertemente asociada con la infección materna12-14. En esta
revisión, destacamos los mecanismos patogénicos moleculares de patógenos
seleccionados que tienen efectos distintos durante el embarazo. El acrónimo TORCH se acuñó para referirse a los patógenos que se sabe que atraviesan la
barrera materno-fetal y causan enfermedades congénitas en el feto15. Además de
los patógenos TORCH tradicionales, también describimos la patogenia de patógenos
emergentes con secuelas importantes para la gestante, el feto y / o el neonato.
Estructura y defensas de la Placenta
Para comprender los mecanismos de transmisión vertical, es fundamental comprender la estructura y función únicas que existen en la interfaz materno-fetal. La placenta humana es un órgano complejo compuesto principalmente por tipos de células especializadas derivadas del feto. dado que las diferencias específicas por edad gestacional en la estructura placentaria y la composición celular se han revisado en detalle en otro lugar16,17, proporcionamos una descripción concisa de la estructura y el desarrollo de la placenta humana a continuación para proporcionar un marco para abordar los mecanismos de patogenia.
En Figura 2a se observa una descripción general de la estructura de la interfaz materno-fetal y en Figura 2b se muestra como placenta humana se desarrolla cuando los trofoblastos derivados del feto, el principal tipo de célula que comprende la placenta, forman el trofectodermo. El trofectodermo forma una barrera celular en las primeras etapas del desarrollo para prevenir la infección del embrión. El proceso de desarrollo placentario continúa rápidamente desde ese punto en adelante y no se completa por completo hasta el final del primer trimestre, sufriendo una serie de cambios morfológicos notables (revisados en referencia 18). Estos cambios dan como resultado el desarrollo de vellosidades coriónicas, que forman el contacto principal entre la placenta derivada del feto y el suministro de sangre materna que eventualmente bañará estas estructuras.
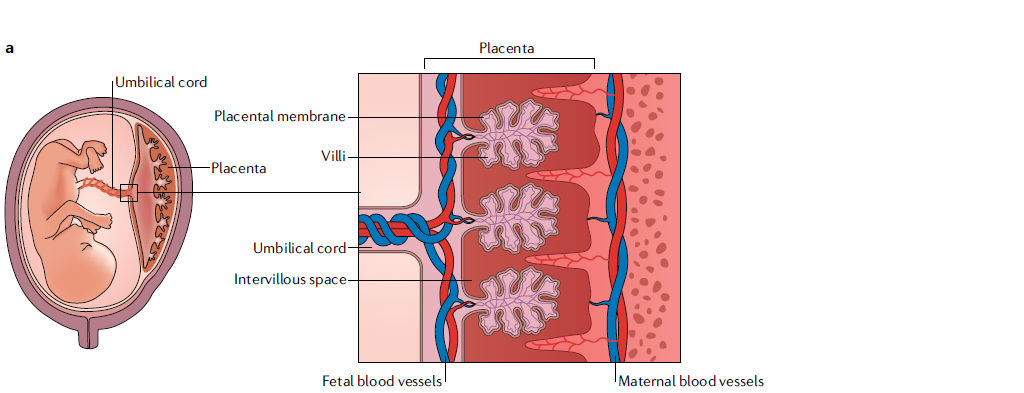
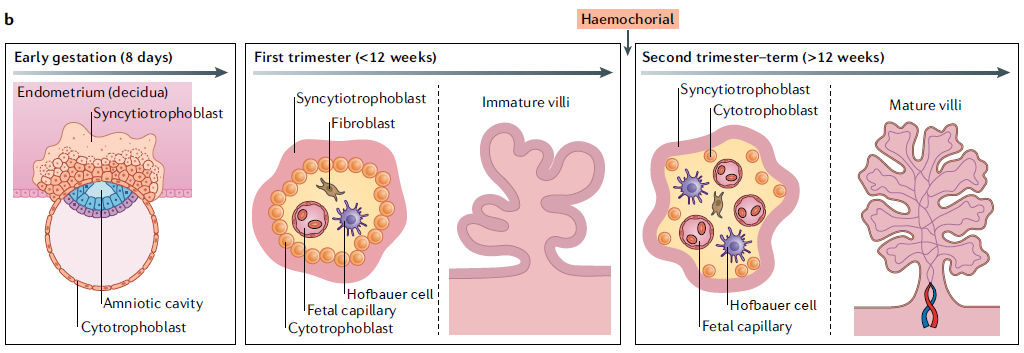
Figura 2.- Estructura y composición celular de la interfaz materno-fetal.
a.- La estructura de la interfaz materno-fetal incluye la decidua materna y la placenta derivada del feto. La microvasculatura uterina materna se remodela para formar arterias espirales, que llevan sangre a las vellosidades coriónicas en el espacio intervelloso.
b.- La placenta sufre una serie de cambios morfológicos rápidos a lo largo de la gestación. Al principio del embarazo (izquierda), el blastocisto se diferencia en el embrión y el trofectodermo, el tipo de célula más temprana que formará la placenta. Los trofoblastos invasores comienzan a invadir la decidua, donde se forma el sincitiotrofoblasto temprano y se infiltra en el endometrio. A lo largo del primer trimestre (al medio de la figura), las vellosidades coriónicas se forman y permanecen inmaduras hasta las últimas etapas de este trimestre. Las vellosidades inmaduras están cubiertas por la capa de sincitiotrofoblasto, con una capa contigua de citotrofoblastos debajo de esta capa. El estroma de las vellosidades en el primer trimestre contiene vasos fetales, que comienzan a formarse alrededor de las seis a ocho semanas de gestación. La microvasculatura materna sufre una remodelación extensa durante el primer trimestre, y la placenta pasa a ser hemocorial al final de esta etapa de la gestación. En el segundo y tercer trimestre (derecha de figura ), las vellosidades coriónicas maduran y permanecen cubiertas por el sincitiotrofoblasto. Sin embargo, a diferencia de las vellosidades inmaduras del primer trimestre, la capa de citotrofoblasto se vuelve discontinua en las últimas etapas de la gestación. En esta etapa, la microvasculatura fetal está completamente desarrollada y el estroma velloso se enriquece en células de Hofbauer derivadas del feto, que se reducen en número más cerca del término completo.
Las células madre de trofoblasto dan
origen a citotrofoblastos (CTBs), las células mononucleares proliferativas de la
placenta y el sincitiotrofoblasto (STB), una capa de células contiguas
multinucleadas que cubre toda la superficie de las vellosidades coriónicas
placentarias. Durante el primer trimestre, la placenta sufre cambios
morfológicos sustanciales que dan como resultado la estructura vellosa que
tendrá durante el resto del embarazo (figura 2b). El final del primer trimestre
también marca la transición crucial a una placenta hemocorial, en la que la
sangre materna contacta directamente con la placenta derivada del feto. Este
proceso requiere trofoblastos extravellosos (EVTs) para remodelar la
microvasculatura materna, lo que finalmente permitirá el suministro de sangre
materna a la superficie de las vellosidades coriónicas placentarias. Este es un
punto crucial ya que discutimos los mecanismos de transmisión vertical
microbiana, que podrían diferir notablemente entre el primer trimestre, cuando
no hay contacto directo entre la placenta y la sangre materna, y las últimas
etapas del embarazo una vez que se establece la placenta hemocorial.
El STB es la principal barrera contra
la propagación hemocorial de agentes infecciosos, particularmente desde el punto
de establecimiento de una placenta hemocorial. Como los CTB proliferativos se
encuentran subyacentes al STB, también se benefician de la protección de esta
capa de células fusionadas. Las vellosidades coriónicas contienen barreras
adicionales a la infección que residen en el estroma velloso, incluyendo
macrófagos derivados del feto (células de Hofbauer) y la microvasculatura fetal,
que deben ser rotos para que un agente infeccioso llegue a la sangre fetal.
Trabajos recientes también sugieren que tipos de células inmunes
derivadas de la madre están muy cerca de la placenta derivada del feto y, por lo
tanto, pueden impartir una capa adicional de protección inmune19. Por lo
tanto, al considerar los mecanismos de transmisión vertical microbiana, es
importante recordar que la transmisión al feto requiere la infección de
múltiples tipos de células y / o la ruptura de las barreras celulares, la más
formidable de las cuales es la STB, como se analiza a continuación. Dado su
papel central en la protección del feto, tal vez no sea sorprendente que la
placenta haya desarrollado mecanismos de acción antimicrobiana altamente
eficientes de defensa. En las siguientes secciones, revisamos los mecanismos
físicos e inmunológicos clave de la defensa placentaria contra la infección.
Defensas físicas
Como se describió anteriormente, el
STB cubre la totalidad de las superficies vellosas coriónicas. El STB contiene
más de 60 mil millones de núcleos y tiene una superficie de 5 m2 en
la mitad de la gestación y de 11 a 12 m2 al término completo20
- una capa celular única aproximadamente del tamaño de un
dormitorio pequeño ! . La propia naturaleza del STB fusionado proporciona
una barrera poderosa para la transmisión microbiana, dado que muchos patógenos
eluden otras barreras celulares debilitando directa o indirectamente las uniones
célula-célula (revisado en Referencia
21). Además de la ausencia total de
uniones celulares, la superficie del STB también representa una barrera para la
adhesión e invasión microbiana dada la presencia de una densa red cortical de
actina subyacente al borde en cepillo (Figura
3).
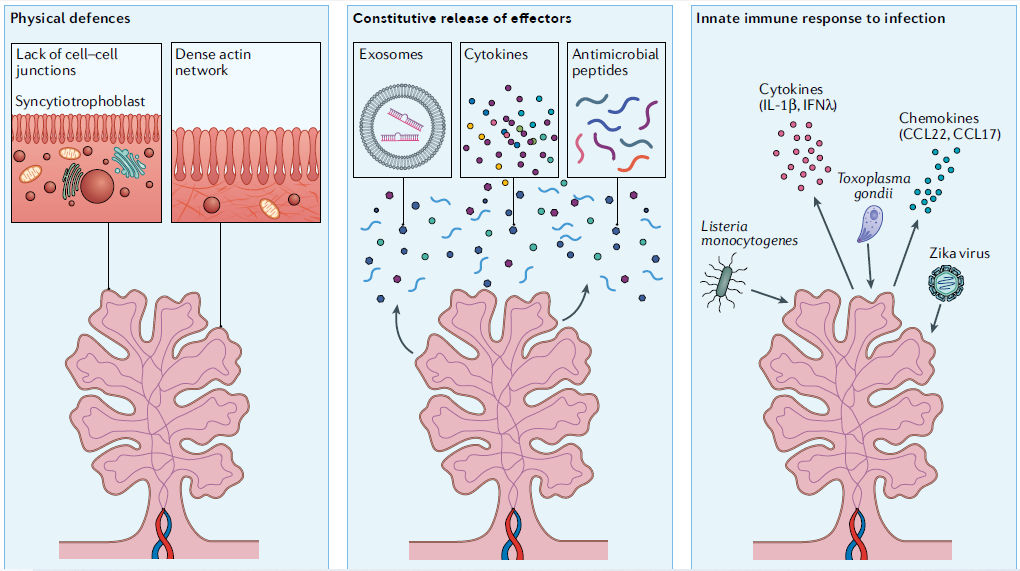
Figura 3.- Defensas placentarias frente a patógenos. Dado su papel como barrera primaria para la diseminación hematógena de agentes infecciosos, la placenta humana ha desarrollado mecanismos dispares y no superpuestos de defensa antimicrobiana. Estos se pueden dividir en al menos tres categorías, defensas físicas (izquierda), liberación constitutiva de efectores antimicrobianos (centro) y / o una sólida respuesta inmune innata a la infección (derecha). Las defensas físicas incluyen la falta de uniones célula-célula de la capa de sincitiotrofoblasto, lo que previene el daño de las uniones intercelulares mediado por la inflamación que podría comprometer la integridad de esta barrera. Las defensas físicas adicionales incluyen la densa red cortical de actina que se encuentra subapical al denso borde en cepillo del sincitiotrofoblasto. Otra forma de defensa implica la liberación constitutiva de potentes efectores antimicrobianos tales como microARNs antivirales en vesículas extracelulares, citocinas (por ejemplo, interferones de tipo III) y péptidos antimicrobianos (al medio). La placenta también responde a los patógenos con una potente señalización inmune innata, que mejora aún más la liberación de sustancias de defensa antimicrobianas (derecha).
Por tanto, la naturaleza física compartida
del STB proporciona mecanismos estructurales comunes para resistir diversos
microorganismos. Sin embargo, cabe señalar que el STB no es la única barrera
celular presente en la placenta humana. El estroma de la placenta también
contiene barreras defensivas, incluyendo la microvasculatura de los vasos
sanguíneos fetales, que también deberían romperse para que los microorganismos
accedan a la sangre fetal. Las defensas físicas de la superficie vellosa
restringen la adhesión y / o la invasión de patógenos no virales, incluyendo
Listeria monocytogenes y T. gondii22-24, pero los virus parecen ser
capaces de entrar en trofoblastos primarios, lo que sugiere que algunos virus
podrían superar las barreras físicas para la entrada de microbios25-28.
Defensas inmunológicas
Decidua.
La superficie de la placenta que mira hacia la madre está en contacto directo con el revestimiento decidual del útero, y esta capa rica en leucocitos es una de las primeras líneas de defensa inmunitaria en la interfase materno-fetal. La decidua materna está en contacto directo y / o indirecto con las membranas fetales, las vellosidades placentarias y la circulación materna. La decidua se puede dividir según estos sitios de contacto: la decidua parietalis recubre la pared uterina; la decidua basal es el sitio de implantación y contacto velloso; y la decidua capsularis recubre la membrana fetal. Las capas deciduales contienen células T citotóxicas, auxiliares (helper) y reguladoras, células asesinas naturales (natural killer o NK), células linfoides innatas, macrófagos y neutrófilos29,30. Hasta el 40% de todas las células inmunitarias deciduales son leucocitos en el primer trimestre y también se observa un infiltrado rico en leucocitos en el parto30-32.
La composición de las poblaciones de
células deciduales muestra una variación específica por edad gestacional y es
importante para la invasión del trofoblasto y para mantener y establecer el
lecho vascular placentario33,34. Como tal, particularmente en el primer
trimestre, las células deciduales permanecen muy próximas a los EVT invasores y
proporcionan defensas inmunes derivadas de la madre en estos sitios (revisado en
referencia 35). A pesar de su composición como un sitio enriquecido con células
inmunes, se ha sugerido que la decidua es un reservorio clave para los patógenos TORCH. El uso de explantes deciduales y los datos de exámenes patológicos de
muestras clínicas sugieren que la decidua puede ser un sitio primario de
replicación para varios patógenos TORCH, incluidos el citomegalovirus (CMV) 36,
el virus Zika (ZIKV) 37 y L. monocytogenes38, y por lo tanto podría formar una
reservorio de patógenos en el sitio clave de la interfaz materno-fetal.
La interacción de la decidua con varios
patógenos in vivo es compleja, ya que los leucocitos deciduales tienen distintas
diferencias fenotípicas de los de la circulación periférica. Los macrófagos
deciduales de origen materno exhiben propiedades únicas en comparación con otros
macrófagos residentes en tejidos o monocitos circulantes sistémicamente19,30,39,40.
Los estudios sugieren que la composición decidual de las células inmunes y su
interacción con los trofoblastos pueden alterar su susceptibilidad a los
patógenos.
Por ejemplo, las células NK deciduales
poseen la notable capacidad de transferir granulisinas a trofoblastos para
proteger de la infección por L. monocytogenes sin matar activamente estas
células41. Estos y otros estudios destacan las estrategias a menudo
únicas y complejas de protección antimicrobiana que existen en la interfaz
materno-fetal. Las células inmunes de la decidua están en contacto con las
células del estroma decidual, las que pueden influir en el comportamiento de las
células inmunes. En particular, la decidua rica en células inmunes ha
alterado la actividad en modelos de cocultivo, lo que refleja que la
arquitectura compleja de la red de células inmunes dentro del estroma decidual
modula la respuesta inflamatoria inmunitaria. Por ejemplo, la función de las
células del estroma decidual se altera con el cocultivo con macrófagos42,
células dendríticas y células NK de una manera dependiente de prostaglandinas43.
Además, la respuesta de las células del estroma decidual a la estimulación de
patógenos es diferente de la de las células del estroma endometrial44-46,
lo que sugiere que el microambiente de la placenta tiene un papel en las
defensas inmunitarias innatas.
Se han utilizado explantes deciduales para demostrar que las células T de memoria efectora CD8 + y las células NK pueden regular las infecciones del estroma decidual por CMV47 y por virus del herpes simple. (VHS) 48 . Estas interacciones multicelulares son difíciles de diseccionar y modelar, pero tienen implicaciones importantes para comprender el panorama inmunológico en la interfaz materno-fetal, ya que cada componente individual (por ejemplo, regulación de células estromales, microambiente hormonal, composición de leucocitos) podría tener un papel en la defensa inmunológica contra la infección.
Vellosidad coriónica.
Las poblaciones de células inmunes fetales en el desarrollo placentario y en patologías asociadas al embarazo se han revisado en otros lugares49,50; por lo tanto, limitamos nuestra discusión en esta sección a estrategias defensivas específicas derivadas del trofoblasto. Además de proporcionar una defensa física contra la infección, también está claro que los trofoblastos vellosos forman una barrera química a la transmisión vertical microbiana. Los trofoblastos tienen una fuerte actividad inmune innata y secretan inmunomoduladores que son importantes para restringir la infección (Figura 3). En el caso de la infección viral, los trofoblastos humanos liberan de forma constitutiva interferones antivirales que restringen la infección tanto de forma autocrina como paracrina51,52, lo que parece ser específico para el STB. La liberación de interferones es una característica muy singular de los trofoblastos, ya que los interferones generalmente solo se inducen en respuesta a la detección de una infección viral. Como los efectores antivirales de la vía del interferón, los genes estimulados por interferón (ISG) pueden ejercer potentes propiedades citotóxicas y proinflamatorias.
Los interferones derivados de trofoblasto son exclusivamente los de la familia de interferones de tipo III: IFNλ1, IFNλ2 e IFNλ3. En ratones, los interferones de tipo III también confieren protección contra infecciones virales, y la deleción del receptor de estos interferones en la placenta sensibiliza al feto a la infección por ZIKV53. La restricción mediada por interferón tipo III de infección en ratones está estrechamente asociada con la edad gestacional, y la mayor protección ocurre después de la placentación53,54. Además, los IFNλ administrados de forma profiláctica o terapéutica en el contexto de la infección por ZIKV en ratones protegen contra la transmisión vertical53,54
Además de los interferones, los trofoblastos humanos también secretan microARN antivirales que se empaquetan en exosomas placentarios, que confieren una amplia protección antiviral en las células receptoras no placentarias y pueden aislarse del suero de mujeres embarazadas25,51,55–57. Dadas estas potentes y no redundantes defensas antivirales, tal vez no sea sorprendente que la mayoría de los virus no puedan replicarse directamente en el STB. Sin embargo, cabe señalar que los efectos de los interferones de tipo I (IFNα e IFNβ) pueden ser distintos de los de los interferones de tipo III. Se ha demostrado que la inducción de la señalización del interferón tipo I materno daña la placenta en ratones in vivo y en explantes de tejido humano ex vivo58. Aunque los mecanismos de estos efectos opuestos aún no se han aclarado por completo, al menos una ISG, proteína transmembrana inducida por interferón (IFITM), inhibe la fusión de CTBs con STB59,60, lo que puede indicar que la expresión de ésta y quizás otras ISGs deben regularse estrictamente en la placenta para evitar daños mediados por interferón. En el caso de la infección bacteriana o parasitaria, muchos estudios destacan la naturaleza resistente del STB, que se ha atribuido en gran medida a las propiedades de barrera física22-24,61
Sin embargo, de manera similar a las
propiedades de la resistencia antiviral, está claro que la placenta
humana también usa vías moleculares adicionales para resistir la infección por
patógenos no virales. Además de la liberación constitutiva de interferones
antivirales, la placenta humana secreta otras citocinas que probablemente
funcionan para limitar las infecciones tanto de manera autocrina como paracrina.
Por ejemplo, la activación constitutiva del inflamasoma en los trofoblastos
placentarios da como resultado la liberación constitutiva de citocinas,
incluidas IL-1β, IL-18 e IL-1α, las que se pueden encontrar circulando sistémicamente en mujeres embarazadas62. Además, la infección de explantes de
placenta humana con L. monocytogenes induce de forma robusta la señalización del
inflamasoma, cuya inhibición sensibiliza a los trofoblastos a la infección62. Al
igual que los efectos de los interferones, los efectos paracrinos de las
citocinas asociadas al inflamasoma también protegen de la infección, ya que la
IL-1β derivada de la placenta prepara las células inmunes circulantes para la
activación posterior del inflamasoma y protege de la infección por L.
monocytogenes62. Por el contrario, la activación del inflamasoma en el
STB
también se ha implicado en una mayor gravedad de los outcomes neonatales
asociados con la malaria placentaria63, lo que sugiere que, de forma similar a
la señalización del interferón, el equilibrio de esta señalización es crucial.
No hay evidencia que sugiera que los
productos secretados derivados de la placenta defiendan directamente contra las
infecciones parasitarias. De hecho, los mismos factores derivados de la placenta
que protegen contra los virus no ejercen ningún efecto sobre la infección por T.
gondii56. En cambio, en el caso de T. gondii, la placenta libera diversos
factores inmunomoduladores, incluidos los quimioatrayentes de células T
reguladoras, quimiocina 22 del motivo C-C (CCL22) y CCL17 en respuesta a la
infección23 (Figura 3). Sin embargo, aún no se ha definido el impacto de esta
señalización en las consecuencias maternas y / o fetales de la infección.
Patógenos TORCH
Como se discutió anteriormente, los
patógenos TORCH constituyen un número limitado de microorganismos con efectos
teratogénicos conocidos. En las siguientes secciones, revisamos los miembros
clave de este grupo que pertenecen a las categorías de bacterias, virus y
parásitos, y las consecuencias de estas infecciones (Tabla
1).
Bacterias
Listeria monocytogenes.
L. monocytogenes es una pequeña bacteria
Gram positiva que se transmite a través de alimentos contaminados. La
listeriosis en el embarazo causa prematuridad y muerte fetal y aumenta la
morbilidad materna con mayores tasas de meningitis y sepsis durante el embarazo
(Tabla 1). En un brote reciente en Sudáfrica, la tasa de mortalidad de los
neonatos fue 28% y de las mujeres embarazadas fue 8% 64. La morbilidad
asociada al embarazo durante este brote fue responsable del 50% de los casos que
se presentaron a la atención de la salud64. A pesar de la inmadurez del sistema
inmunológico fetal al comienzo del embarazo, se ha demostrado que el tratamiento
de L. monocytogenes en el primer trimestre después de la exposición mejora los
resultados fetales65–67. Por el contrario, no se ha demostrado definitivamente
que el tratamiento después del primer trimestre cambie el curso de la enfermedad
en el feto, pero se recomienda para mejorar los resultados maternos68. Esto
probablemente se deba a una mayor susceptibilidad del feto a la infección por L. monocytogenes después del primer trimestre y sugiere que existen diferencias
específicas por edad gestacional en la transmisión vertical. Después de entrar
a través de la mucosa entérica, L. monocytogenes se propaga de célula a célula
manipulando la polimerización de actina.
Se desconoce el mecanismo de transmisión transplacentaria de la bacteria. En
cultivos ex vivo, el STB es altamente resistente a la infección24,62. Los
factores de virulencia bacteriana necesarios para entrar en otras células (por
ejemplo, listeriolisina O e internalina B) no son necesarios para entrar en
líneas celulares de trofoblasto de coriocarcinoma humano69. La proteína
bacteriana internalina InlA es importante para unirse a E-cadherina en
trofoblastos primarios70, pero su papel en el cruce de la barrera placentaria in
vivo sigue sin estar claro, con efectos diferenciales en diferentes modelos
animales71,72. Juntos, estos datos sugieren que la transmisión a través de la
barrera placentaria puede requerir factores de virulencia bacteriana
adicionales, aunque no caracterizados. En consecuencia, hubo una variación
mínima de la cepa con respecto a la capacidad de infectar la placenta en
estudios que utilizaron un modelo competitivo in vivo en cobayas73, lo que
sugiere que los factores de virulencia específicos del trofoblasto se conservan
en las cepas epidémicas
Después de la colonización placentaria, la bacteria se disemina a los tejidos fetales y se puede resembrar nuevamente a los órganos maternos74. En ratones, la polimerización de actina a través de la proteína bacteriana ActA se ha demostrado que es importante para la diseminación de célula a célula y transmisión a través de la interfaz materno-fetal75
El factor de colonización bacteriano secretado internalina P es importante para distorsionar las células y permitir que las bacterias atraviesen la membrana basal76, pero aún no se han caracterizado los mecanismos de transporte a través del STB a la membrana basal. Es importante destacar que la infección fetal se asocia con el desarrollo de abscesos placentarios y el reclutamiento de células inmunes innatas en la interfaz materno-fetal77. Los estudios en modelos de primates no humanos demuestran que no se requiere que la bacteria atraviese la barrera materno-fetal para causar inflamación placentaria, pérdida y compromiso fetal 78, lo que sugiere que la pérdida del embarazo también puede ocurrir en ausencia de colonización bacteriana del feto.
Treponema pallidum.
T. pallidum es un teratógeno bien
caracterizado. Esta bacteria tiene una longitud de 6 a 20 μm, tiene un genoma
pequeño de solo 1.041 marcos de lectura abiertos y una membrana externa que
cubre una capa de peptidoglicano, pero con un mínimo de proteínas expuestas a la
superficie y sin lipopolisacárido79. La falta de proteínas de superficie puede
facilitar la evasión de la respuesta inmune y la notable longevidad de la
bacteria en el huésped humano79. T. pallidum puede colonizar y causar
enfermedades en todos los sistemas de órganos fetales y es el agente causante de
la sífilis congénita (Tabla 1). Más del 50% de las mujeres con sífilis tienen
resultados adversos en el embarazo80-82, y la transmisión fetal causa una amplia
variedad de patologías neonatales que incluyen meningitis, osteocondritis,
supresión de la médula ósea e hidrops83-85 (tabla
1). La sífilis congénita está en
aumento en los Estados Unidos, con 1.306 casos reportados en 2018 (referencia
86) y es una de las principales causas de pérdida fetal mediada por infección y
morbilidad neonatal en el mundo en desarrollo, causando un estimado de 150.000
mortinatos, 60.000 muertes neonatales y 100.000 infecciones de lactantes al año81,82.
La fisiopatología de la transmisión congénita de T. pallidum sigue siendo en
gran parte desconocida. Las secuelas del feto con sífilis congénita dependen
tanto del estadio materno de la infección como de la edad gestacional del
feto84,85. La transmisión congénita ocurre con menos frecuencia en embarazos
posteriores, una observación conocida como ley de Kassowitz. Esta observación
sugiere que la respuesta inmune materna a T. pallidum puede limitar la
frecuencia de la sífilis congénita, pero el riesgo de transmisión vertical nunca
se elimina87–90. Esto también se ha demostrado en un modelo de sífilis congénita
en cobaya, que es el modelo mejor descrito para este fenómeno90. Además, en el
modelo de cobaya, la respuesta de IgM fetal se corresponde más con la
transmisión vertical que con el ADN espiroquetal91, lo que sugiere que la
respuesta inmune fetal es importante en la patogénesis de la sífilis congénita.
Se ha demostrado inflamación y maceración
del tejido placentario en casos de pérdida del embarazo o muerte fetal
intrauterina asociada con sífilis congénita en ausencia de anomalías congénitas,
lo que sugiere que la tasa de pérdida fetal de hasta 50% asociada con la
sífilis materna podría atribuirse a la respuesta inflamatoria. en la propia
placenta92,93. Junto con esto, la reacción de Jarisch-Herxheimer (respuestas
inflamatorias intensificadas que ocurren con el tratamiento con antibióticos de
la
sífilis en el embarazo) se asocia con contracciones prematuras y evidencia de
insuficiencia uteroplacentaria94, lo que sugiere que la inflamación placentaria
tiene un papel en la patogenia del parto prematuro asociado con la sífilis.
Otras bacterias
Como se mencionó anteriormente, la placenta forma una barrera que restringe eficazmente muchos patógenos bacterianos. Aunque hay evidencia de que la inflamación sistémica puede causar un parto prematuro y la fiebre en sí misma puede ser teratogénica, no hay evidencia de que otros patógenos bacterianos (por ejemplo, especies de Klebsiella, Staphylococcus aureus resistente a la meticilina (MRSA), Escherichia coli) atraviesen rutinariamente la barrera placentaria y causen infección fetal. Estos patógenos son causas importantes de morbilidad materna en el contexto de una infección materna sistémica (por ejemplo, pielonefritis), pero no son causas importantes de infección fetal, lo que sugiere que la barrera materno-fetal frente a múltiples patógenos bacterianos permanece intacta en el caso de la enfermedad sistémica materna. enfermedad y bacteraemia. Los mecanismos que subyacen a las defensas antibacterianas de la placenta en el contexto de la bacteriemia siguen sin estar caracterizados en gran medida.
Por el contrario, existe un subconjunto de patógenos bacterianos bien descritos que ascienden por el tracto genital y causan corioamnionitis (revisado en referencias 95,96). La infección intraamniótica ocurre clásicamente en el contexto de la rotura de membranas, lo que sugiere que el puente de la membrana fetal proporciona una barrera importante para la infección fetal. En cultivos placentarios y neonatales se han encontrado patógenos endémicos de la microflora vaginal tales como estreptococo del grupo B (GBS), E. coli y especies de Bacteroides97. Tanto los modelos de primates como los de murinos demuestran que los microorganismos vaginales pueden atravesar el tracto genital y causar una enfermedad diseminada en la cavidad fetal. Típicamente, las infecciones surgen de la microbiota genitourinaria y pueden ser polimicrobianas97,98. Para causar una enfermedad en la cavidad uterina, estas bacterias tendrían que ascender por la biopelícula microbiana vaginal y el tapón mucoso cervical antimicrobiano para entrar en contacto con las membranas fetales y causar una respuesta inflamatoria local.
Estreptococo Grupo B
El microorganismo mejor estudiado asociado con la infección ascendente y la enfermedad neonatal es el EGB , que puede causar sepsis neonatal con transmisión. El screening de colonización y la profilaxis intraparto con antibióticos disminuye la incidencia de sepsis neonatal de inicio temprano 99. Se han observado biopelículas de EGB en las membranas fetales100 y es una de las bacterias más comúnmente cultivadas en el contexto de infecciones intraamnióticas97. El EGB coloniza el tracto genital en ~ 10 a 30% de las mujeres101. La predilección de algunos aislados por causar enfermedad neonatal sigue sin estar clara, pero el EGB activa la respuesta inmune innata de neutrófilos y macrófagos para iniciar la liberación de citocinas, la migración celular y el desarrollo de trampas extracelulares y bacteriolisis102,103. Los mecanismos de cambio patogénico y determinantes de virulencia específicos para ascender por el tracto genital incluyen pili, adhesinas y sistemas reguladores alterados (revisado en las referencias 104,105).
E. Coli
E. coli también puede adherirse a las membranas fetales, aunque en menor grado que el EGB106. E. coli, como EGB , habita en el tracto genital y gastrointestinal y comúnmente se aísla en el contexto de una infección intraamniótica. Con la implementación del screening universal y la profilaxis intraparto con antibióticos para el SGB, E. coli se ha convertido en el agente etiológico más común de la sepsis de inicio tardío en los recién nacidos97,107. En los aislados obstétricos, los subgrupos filogenéticos son variables, pero los genes de las islas de patogenicidad para las fimbrias de tipo 1 y la adquisición de hierro están enriquecidos108, lo que sugiere que la adquisición horizontal de genes es importante para causar enfermedades durante el embarazo. Al igual que con otros patógenos bacterianos, los factores de virulencia específicos que permiten que E. coli cause una enfermedad clínica y asciendan por el tracto reproductivo durante el embarazo siguen siendo objeto de estudio.
Virus
Tanto los virus de ADN como de ARN pueden
atravesar la interfaz materno-fetal y causar enfermedad fetal. En las siguientes
secciones, discutimos los virus teratogénicos clave y lo que se conoce con
respecto a sus rutas de transmisión transplacentaria y resultados clínicos.
Aunque el virus de la rubéola era una fuente importante de enfermedad congénita
antes de los esfuerzos de vacunación exitosos, hemos restringido nuestra
discusión a los virus más comunes actualmente asociados con la enfermedad
congénita contemporánea: CMV, HSV1 y HSV2, parvovirus B19, VIH y ZIKV (Tabla 1)
Citomegalovirus.
El CMV es un miembro de la familia Herpesviridae y es una de las causas más comunes de infecciones verticales a nivel mundial. Es la causa más común de pérdida auditiva congénita en los Estados Unidos, y la carga mundial de morbilidad también es alta, aunque probablemente subestimada109. Como otros virus del herpes, CMV entra en un estado latente después de una infección aguda y puede reactivarse posteriormente. En el contexto de una infección primaria materna, existe un riesgo de transmisión fetal de ~ 40%. Por el contrario, el riesgo de transmisión al feto después de la reactivación es < 0,05%, pero causa la mayor parte de la enfermedad clínica110,111. Se han descrito variaciones específicas según edad gestacional en la infectividad y consecuencias fetales112, lo que sugiere que las alteraciones en la interfaz materno-fetal durante la gestación regulan los mecanismos de la infección vertical por CMV. La histología placentaria en la infección congénita por CMV varía de normal a aguda e intervilositis crónica113. Los cuerpos de inclusión virales clásicos del ojo de búho ocurren con más frecuencia en el primer y segundo trimestres que en la placenta a término114.
Dado que otras revisiones recientes han proporcionado descripciones detalladas de los mecanismos por los cuales el CMV podría transmitirse verticalmente115-117, aquí proporcionamos un descripción general más centrada. Se ha demostrado que el CMV infecta a los trofoblastos, pero con diferencias sustanciales de infectividad relacionadas con la edad gestacional28. Dado que el CMV humano no infecta fácilmente a los roedores, los estudios in vivo de los mecanismos de transmisión vertical del CMV se limitan a cultivos de células primarias placentarias humanas y / o modelos de explantes. Aunque se han descrito modelos de cobayas de infecciones por CMV durante el embarazo118, estos no recapitulan completamente el fenotipo de las infecciones humanas por CMV durante el embarazo.
Los estudios de la infección por CMV en tejido humano demuestran una replicación activa en la decidua de origen materno, lo que puede generar un reservorio viral que podría aumentar la probabilidad de que el CMV cruce la barrera fetal en el contexto de una infección primaria36,119. Específicamente, la enzima de edición de ARNm de apolipoproteína B 3A de tipo polipéptido catalítico (APOBEC3A) se ha implicado en el control de la replicación del CMV en la decidua, pero no en las vellosidades placentarias37. Los estudios también han demostrado que el CMV puede infectar preferentemente a los pericitos placentarios para acceder al feto y que este podría ser un modo primario de infección en el contexto de la viremia materna120. El papel que tienen estas células en la interfase materno-fetal permanece en gran parte sin caracterizar, pero pueden representar un sitio importante desde el cual el CMV puede atravesar la placenta y causar una infección congénita, particularmente con el avance de la gestación
Herpes simplex virus.
El VHS1 y el VHS2 pueden transmitirse al feto por vía transplacentaria, pero la transmisión a través del contacto con una lesión de diseminación del virus en el tracto genital es un modo de transmisión vertical mucho más común. HSV1 y HSV2 son neurotrópicos y permanecen latentes en el ganglio de la raíz dorsal después de la infección primaria. Al llegar al feto, estos virus son altamente teratogénicos y se asocian a la clásica tríada de manifestaciones en piel (aplasia cutis, cicatrización, erosiones), sistema nervioso central (ventriculomegalia, microcefalia, calcificaciones intracraneales) y ojos (coriorretinitis, atrofia), junto con con manifestaciones esqueléticas y pérdida fetal.
Un modelo de ratón recapitula la transmisión vertical, con pérdida fetal, malformaciones congénitas y neurotropismo, demostrando que la diseminación hematógena (más bien que una infección ascendente) es responsable de los efectos sobre el feto121. Los mecanismos de transferencia transplacentaria de HSV1 y HSV2 no están caracterizados en gran medida. El STB es resistente a la infección por HSV1 y HSV2, pero los EVTs son permisivos25,122. Aunque los mediadores de entrada de HSV HveA, HveB y HveC se expresan en EVTs, los anticuerpos contra HveA122 no bloquean la transmisión viral. La superficie materna de la placenta es positiva para HSV1 o HSV2 en el 9-28% de las mujeres que están asintomáticas en el momento del parto sin evidencia de transmisión fetal123,124, lo que sugiere que la placenta y / o la respuesta inmune materna presentan una barrera contra la placenta transplacentaria. transmisión en mujeres con infección no primaria.
Parvovirus B19.
El parvovirus B19 es un virus de ADN
monocatenario sin envoltura de la familia Parvoviridae. La infección por
parvovirus B19 es común durante la niñez y causa fiebre baja, exantema
maculopapular y exantema en la mejilla. La inmunidad humoral protege contra las
infecciones y el 70% de la población adulta es inmune125. La tasa de transmisión
de la infección materna por parvovirus B19 al feto es 17 - 33% y la mayoría
de los fetos tienen una resolución espontánea sin secuelas, aunque ~ 3% puede
desarrollar hidrops no inmune por anemia fetal9. La pérdida fetal y la muerte
fetal se asocian con infección incluso en ausencia de hidrops. Las secuelas
de la infección varían con la edad gestacional, de manera que la infección
después de las 20 semanas confiere un riesgo de pérdida fetal del 0,5%, mientras
que antes de las 20 semanas la tasa aumenta en 30 veces 9.
El parvovirus B19 exhibe un fuerte tropismo por los precursores eritroides y la
inmunohistoquímica demuestra que después de atravesar la placenta el virus
alcanza el endotelio fetal 126. Se ha demostrado que la proteína de la cápside
del parvovirus B19 VP2 se une al globoside, que está presente en las superficies
de los STB y CTB127. También se ha demostrado que la proteína de la cápside VP2
se une a las células del trofoblasto velloso a través de glicolípidos
globosidos128. Después de la entrada del virus, la proteína no estructural 1
(NS1) induce la apoptosis en las células129. De acuerdo con esto, la apoptosis
también se ha demostrado en la placenta en embarazos gravemente afectados, lo
que sugiere que la muerte fetal en ausencia de hidropesía puede resultar de daño
placentario que afecta directamente la función130. La secuela rara pero clásica
del parvovirus B19 es la hidropesía fetal no inmune causada por una anemia grave
y transitoria con pérdida tanto de eritrocitos como de precursores eritroides
nucleados129. Se ha informado discordancia de gemelos dicoriónicos en la
infección y en las secuelas clínicas131,132, lo que sugiere que las respuestas
placentaria y fetal son importantes para limitar la enfermedad clínica, pero los
mecanismos inmunes por los que esto ocurre no están claros.
HIV.
La transmisión congénita del VIH permanece asociada con la morbilidad neonatal global. El VIH se puede transmitir verticalmente a través de la ruta transplacentaria, durante el parto y / o durante la lactancia después del nacimiento. En ausencia de terapia antirretroviral (TAR), ~ 25% de los bebés nacidos de mujeres VIH positivas se infectan en comparación con < 2% de aquellos cuyas madres están en TAR133. Las contribuciones relativas de las diversas rutas de transmisión vertical sugieren que cada una tiene un papel en la infección congénita por VIH, pero la mayoría de las infecciones congénitas ocurren con transmisión intraparto y exposición directa a secreciones y sangre maternas.
Ex vivo, explantes de vellosidades coriónicas aislados de placentas del primer trimestre y a término completo apoyan la infección por VIH, sugiriendo la posibilidad de que la placenta pueda infectarse durante el embarazo134,135. Sin embargo, aunque las proteínas y / o los ácidos nucleicos del VIH pueden detectarse en el tejido placentario recolectado de mujeres VIH positivas, esto ocurre con relativa poca frecuencia, y los estudios demuestran que la mayoría de placentas recolectadas de mujeres VIH positivas son negativas para el VIH136-138. Estos datos coinciden con los datos epidemiológicos que sugieren que, aunque es posible la transmisión transplacentaria del VIH, es una ocurrencia poco frecuente (< 1% )139,140. Sin embargo, la coinfección materna con otros patógenos como el CMV o la malaria puede aumentar aún más el riesgo de transmisión vertical y / o outcomes adversos. (revisado en referencia 141).
Zika virus.
El ZIKV es un virus de ARN de cadena positiva de la familia Flaviviridae que provocó un gran brote de más de 2000 casos de enfermedad congénita en 2015-2016, con epicentro en Brasil142,143. El ZIKV se transmite a través de vectores dependientes (mosquitos Aedes aegypti) y vías independientes de vectores (por ejemplo, transmisión sexual , por transfusión de sangre y transmisión vertical). Las infecciones maternas por ZIKV se asocian con una variedad de outcomes clínicos fetales que incluyen ventriculomegalia, microcefalia y retrasos en el desarrollo (Tabla 1), que ocurren en ~ 10% de los pacientes con infección materna144. La discrepancia entre la infección fetal y las diversas secuelas neurológicas de la enfermedad ha llevado a la separación de la infección congénita por ZIKV y el síndrome congénito por ZIKV145. Desde el brote inicial, los casos de enfermedad congénita por ZIKV han disminuido considerablemente, lo que sugiere que la inmunidad preexistente es importante para regular la infección por ZIKV del huésped materno. La infección por ZIKV en explantes de placenta humana y modelos de ratón sugiere claras diferencias de edad gestacional en la eficacia de la transmisión y las secuelas fetales, siendo la infección en el primer trimestre el que presenta el mayor riesgo37,53,144.
Interesantemente, se ha demostrado discordancia en gemelos para infecciones placentarias y neonatales con hasta un 50% de discordancia en pares de gemelos146, y esta susceptibilidad puede recapitularse in vitro después del parto147, lo que sugiere que los factores derivados de la placenta o el feto son importantes para limitar la transmisión vertical. Al igual que otros patógenos TORCH, los mecanismos por los cuales el ZIKV atraviesa la barrera placentaria siguen siendo en gran parte desconocidos (Figura 4), a pesar de una investigación considerable. Algunos estudios han asociado el tropismo de las células placentarias de la infección por ZIKV con la expresión del receptor, pero los receptores que son importantes en la unión de flavivirus (por ejemplo, la familia de receptores TAM) no lo son para la infección por ZIKV in vivo148. Los estudios in vitro en ratones y ex vivo en humanos también han sugerido que los anticuerpos preexistentes contra el virus del flavivirus dengue relacionado (DENV), que es co-endémico con ZIKV, potencia la infección congénita por ZIKV149,150 y facilita el cruce viral a través de la placenta.
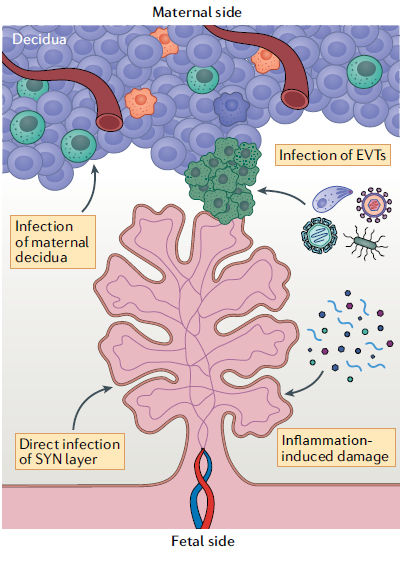
Figura 4.- Posibles mecanismos de transmisión vertical. Los mecanismos por los cuales muchos patógenos TORCH (Toxoplasma gondii, otros, virus de la rubéola, citomegalovirus, virus del herpes simple) acceden al feto no están claros. Sin embargo, los estudios sugieren que algunos de estos patógenos pueden utilizar vías similares para eludir la barrera placentaria. Estos mecanismos incluyen la infección de trofoblastos extravellosos (EVTscélulas verdes) y / o la infección de la decidua derivada de la madre, como por ejemplo a través de la infección directa de poblaciones de células inmunitarias maternas. Otras rutas posibles incluyen la infección directa de la transmisión a través de las vellosidades coriónicas, a través de la infección directa del sincitio (SYN) o mediante el daño mediado por inflamación de la capa de sincitiotrofoblasto que rompe la barrera y permite la transmisión.
Sin embargo, los datos de estudios en humanos en regiones endémicas sugieren que
la inmunidad preexistente al DENV está asociada con un riesgo reducido de
infección por ZIKV151,152, lo que está respaldado por el trabajo en modelos de
primates no humanos153,154. Por el contrario, la preexposición al ZIKV
sensibiliza al riesgo subsiguiente de DENV grave en humanos155. Los modelos de
primates no humanos de transmisión vertical del ZIKV han proporcionado
importantes conocimientos sobre la naturaleza compleja de las infecciones
materno-fetales con este virus (revisado en la referencia 156) y también han permitido
estudios que investigan las secuelas neonatales e infantiles157. Sin embargo,
incluso estos modelos aún tienen que dilucidar los mecanismos precisos por los
cuales el virus atraviesa la barrera placentaria. Tanto los modelos animales
como los in vitro destacan la complejidad multifactorial de la transmisión
vertical del ZIKV y sugieren que el modelado de este fenómeno puede ser difícil
Virus emergentes
Varios virus emergentes pueden tener un impacto importante en la madre y el feto durante el embarazo. Aunque el embarazo a menudo no está bien estudiado en el contexto de pandemias emergentes, hay datos que indican una mayor gravedad de la infección materna por el virus del Ébola (EBOV) y reportes de transmisión vertical del virus de la fiebre del Valle del Rift (RVFV), síndrome respiratorio agudo severo coronavirus 2 ( SARS-CoV-2), virus del Nilo Occidental (VNO) y virus de la encefalitis equina del este (EEEV).
EBOV
El EBOV pertenece a la familia Filoviridae, que son virus de ARN con envoltura de cadena negativa. Las infecciones por EBOV se asocian con hemorragia materna, trabajo de parto prematuro, aborto espontáneo y muerte materna y fetal. La muerte materna ocurre en el 85% de los casos con casi el 100% de pérdida de la descendencia (muerte fetal, aborto espontáneo y muerte neonatal) 158. Además de causar una enfermedad materna grave, también hay informes que sugieren la transmisión vertical de EBOV.
Durante el brote de 2013-2016 en África occidental se observó que mujeres embarazadas sobrevivían a la enfermedad por EBOV incluso con viremia clara, pero dieron a luz bebés muertos con niveles elevados de ARN viral en hisopados de placenta y feto hisopos, lo que sugiere una transmisión vertical de EBOV159,160. Además, el antígeno EBOV se ha encontrado en el STB por inmunohistoquímica, aunque esta localización por sí sola es insuficiente para demostrar la transmisión vertical y, en cambio, puede sugerir que el STB restringió con éxito el acceso a la sangre fetal159
RVFV
El RVFV es un arbovirus de la familia
Bunyaviridae que se transmite a través de un vector artrópodo y está asociado
con una enfermedad grave en animales domésticos y ganado. En los rebaños, los
brotes de RVFV se asocian con "tormentas de abortos", en las que se observan
pérdidas fetales y / o mortinatos en hasta un 90-100% de las hembras preñadas.
Un estudio de caso único ha demostrado la transmisión vertical en el tercer
trimestre en humanos, y también se ha descrito un aumento de los abortos
espontáneos y la muerte fetal en asociación con RVFV161,162. En tejido
placentario humano ex vivo, se ha demostrado la replicación viral tanto en el STB como en los CTBs163,164, pero la ruta específica de transmisión vertical en
el ganado y en los humanos, y si estas rutas son similares, sigue sin estar
clara.
WNV
El WNV es un flavivirus transmitido por artrópodos asociado con una patología neurotrópica marcada. Ha habido varios informes de casos de transmisión vertical en humanos, y una serie de casos demostró una asociación con anomalías del sistema nervioso central (hidrocefalia, microcefalia y lisencefalia) 165, pero se necesitan estudios de casos más amplios para confirmar la asociación y la frecuencia de la transmisión vertical permanece desconocida. En modelos de ratón, el WNV induce muerte y enfermedad fetal similar al ZIKV, lo que sugiere un potencial para causar enfermedad congénita in vivo166. Además, el tejido de explante humano, incluyendo muestras de decidua, vellosidades coriónicas y membranas fetales, es permisivo en diversos grados166. En las vellosidades placentarias, los EVTs son infectados preferentemente166 y el STB sigue siendo en gran parte resistente a la infección166,167, similar a lo que se describe para la infección por ZIKV.
Hallazgos similares con el virus Powassan (POWV) sugieren que algunos arbovirus neurotrópicos emergentes tienen el potencial de afectar el desarrollo fetal si se transmiten verticalmente135. Por el contrario, otros arbovirus, incluidos el virus chikungunya (CHIKV) y el virus Mayaro (MAYV), no causaron enfermedad fetal en ratones y se replicaron de manera ineficaz en explantes de placenta humana, lo que sugiere diferencias en el potencial teratogénico de los arbovirus emergentes135.
Sars- Cov - 2
Más recientemente, la pandemia de SARS-CoV-2 ha puesto de relieve la necesidad de centrarse en la salud de la mujer, especialmente durante el embarazo. Varios informes de casos de mujeres embarazadas con COVID-19- describen la infección del STB por inmunohistoquímica acompañada de lesiones patológicas placentarias168,169. Es importante destacar que la gran mayoría de los casos no muestran ninguna evidencia de transmisión vertical170-173, lo que sugiere que la placenta conserva su función de barrera incluso en el contexto de una infección y enfermedad maternas graves. Por lo tanto, a pesar de la magnitud de la pandemia, el riesgo para el feto como resultado de la transmisión vertical del SARS-CoV-2 parece ser mínimo y no hay datos que sugieran que el SARS-CoV-2 sea de alguna manera teratogénico por sí mismo.
Parásitos
Hay varias infecciones parasitarias
relevantes en el embarazo que causan enfermedades fetales a nivel mundial. Estos
parásitos difieren en el modo de transmisión al anfitrión materno, pero todos
tienen implicaciones sustanciales para la salud maternoinfantil mundial.
Toxoplasma gondii.
Hay más de 200.000 casos de toxoplasmosis congénita en todo el mundo cada año174. T. gondii infecta cerca de 6 a 70% de los fetos con la infección materna dependiendo de la edad gestacional en el momento de la infección, y puede tener consecuencias devastadoras, como ventriculomegalia, calcificaciones intracraneales, coriorretinitis y rara vez hidrocefalia174-176. Los síntomas maternos ocurren solo en ~ 5% de los casos y, por lo tanto, las secuelas clínicas se caracterizan mejor en estudios en los que se realizan screening prenatales de rutina, ya que la infección materna es subclínica. Los fetos infectados al principio del embarazo tienen muchas más probabilidades de presentar una enfermedad clínica. Solo el 9% de las mujeres con seroconversión al final del embarazo dan a luz niños con retrasos en el desarrollo neurológico (en comparación con el 25% de las que presentaron seroconversión en el segundo trimestre) 177.
La infección por T. gondii se ha estudiado
ampliamente a nivel celular y molecular en muchas células no placentarias. La
infección es un proceso de varios pasos que incluye la adhesión entre las
moléculas de superficie de T. gondii y los proteoglicanos de la superficie de la
célula huésped, la adhesión impulsada por la secreción de proteínas en la
membrana plasmática del huésped y la invasión a la célula huésped mediada por
interacciones entre las proteínas de la superficie del parásito y
proteínas del parásito secretadas sobre la superficie celular (revisadas en las referencias
178,179). T. gondii también secreta efectores proteícos en la célula huésped
durante la infección (revisado en referencias 180, 181). Estos efectores
diversos alteran muchos aspectos fundamentales de la biología de la célula
huésped, incluyendo las vías inmunes innatas que son importantes para la
señalización defensiva181. A pesar de la caracterización en células no
placentarias, los mecanismos de transmisión vertical de T. gondii siguen sin
estar claros.
El STB muestra una clara resistencia a la infección por T. gondii, la que se
produce a nivel de adhesión y replicación posterior a la entrada23,24,61. Por el
contrario, los CTBs y EVTs no muestran el mismo grado de resistencia23,24, lo
que sugiere diferencias específicas de tipo celular en los mecanismos de
resistencia. Además de las defensas intrínsecas de las células, los trofoblastos
también responden a la infección por T. gondii mediante la inducción específica
de diversas citocinas y quimiocinas, incluyendo la inducción robusta de la quimiocina reguladora de células T CCL22 (referencia 23).
Remarcablemente , esta
inducción no está impulsada por la detección del trofoblasto, sino que requiere
la administración del efector molecular GRA28 derivado de T.gondii-
(referencias 23,182). Sin embargo, no está claro qué papel tienen las citocinas o
quimiocinas derivadas de la placenta en la patogénesis y la transmisión vertical
de T. gondii
Plasmodium especies.
De las cuatro especies de paludismo o
malaria que
causan enfermedades en los seres humanos, Plasmodium falciparum causa la mayoría
de las enfermedades durante el embarazo. En áreas de baja transmisión y baja
inmunidad preexistente, la infección congénita es sintomática y causa paludismo
cerebral, síndrome de dificultad respiratoria, hipoglucemia refractaria y aborto
espontáneo y mortinato. En áreas donde el parásito es endémico y la inmunidad
preexistente es alta, la malaria puede causar anemia materna grave, parto
prematuro y restricción del crecimiento fetal, y sigue siendo una de las
principales causas de mortalidad neonatal183-185. Se ha informado de infección
congénita y los datos sugieren que la adquisición perinatal a través de la
transmisión vertical también ocurre en áreas endémicas186-188.
El parásito Plasmodium tiene una clara predilección por la placenta. La unión de
los eritrocitos infectados a la placenta permite al parásito evadir la respuesta
inmune materna cambiando los antígenos de superficie (que se describen en
detalle a continuación), y las muestras histopatológicas de la infección
patógena por paludismo contienen una gran cantidad de eritrocitos en el espacio
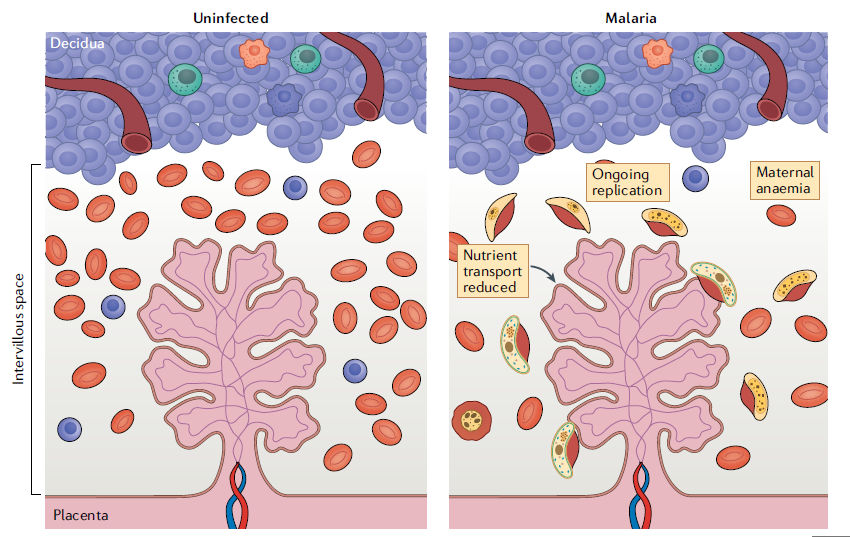
intervelloso con parásitos e intervellositis agudas y crónicas. 189,190. Debido a la carga de malaria placentaria, el transporte
de nutrientes a través de la placenta se reduce, lo que pone de relieve cómo la
función placentaria puede verse comprometida por la infección191,192 (Figura 5).
Los mecanismos para la unión específica a placenta y el secuestro de los
eritrocitos infectados se ha caracterizado relativamente bien. Los eritrocitos
infectados con Plasmodium se unen a receptores endoteliales para el secuestro
tisular en tejidos no placentarios a través de antígenos de superficie variantes
codificados por parásitos (VSAs) que son expresados por el eritrocito infectado
(revisado en la referencia 193).
La expresión de la familia de genes VSA "var" varía durante el curso de una infección. var codifica ~ 60 proteínas de la superficie de los eritrocitos anotadas la proteína 1 de la membrana de los eritrocitos de P. falciparum (PfEMP1), que son responsables de la adhesión a eritrocitos no infectados y otros tipos de células193-195. Durante el curso de la infección, se produce una variación antigénica en la expresión de diferentes miembros de la familia en la familia de genes var, lo que permite la evasión inmunitaria.
Durante el embarazo, el parásito se adapta a la presencia de la
placenta, permitiendo el secuestro placentario y la evasión inmunitaria, así
como tambien la enfermedad materna y fetal (Figura 5). El gen de Plasmodium que codifica
VSA 2CSA (var2csa) se transcribe selectivamente en el paludismo placentario y
permite que el eritrocito infectado se adhiera al STB195-197. Fuera del
embarazo, el CD36 es un receptor principal para los eritrocitos infectados, pero
el condroitín sulfato A (CSA) en el STB se convierte en el principal receptor de
VAR2CSA y es importante para el secuestro placentario198-200. La regulación
positiva de VARCS2A por parte del parásito en eritrocitos infectados durante el
embarazo permite la evasión de una respuesta inmune preexistente en áreas
endémicas y explica cómo la primigravidez (es decir, estar embarazada por
primera vez) es un factor de riesgo de enfermedad grave.
En consecuencia, los anticuerpos contra los VSAs específicos de la placenta,
incluyendo VAR2CSA, no están presentes en los hombres y no se desarrollan hasta
más tarde en el embarazo201-203. Los VSA son los objetivos principales de la IgG
materna, y los niveles elevados y una mayor afinidad por los VSA protegen de la
enfermedad grave204-206. El secuestro placentario de eritrocitos infectados
causa afluencia de fagocitos y citocinas, que se cree dañan el STB
adyacente y conduce a resultados adversos en ausencia de parasitemia
fetal194,207,208 (Figura 5). Recientemente, se ha demostrado que la autofagia
placentaria, un mecanismo para mantener la homeostasis celular normal, está
desregulada en la infección por malaria placentaria, lo que causa
disminución del transporte de aminoácidos al feto y se ha implicado en el
desarrollo de la restricción del crecimiento fetal 209,210.
Figura 5.- Paludismo placentario en zonas endémicas. Esquema de la interfaz materno-fetal en estado no infectado (panel izquierdo). Esquema de la interfaz materno-fetal en el contexto de la malaria (panel derecho). El parásito (amarillo) accede a la placenta a través de la circulación materna. El espacio intervelloso luego se convierte en un sitio de replicación del parásito donde el parásito sufre una variación antigénica de superficie, lo que finalmente conduce a un escape inmunológico y una mayor replicación que causa anemia materna. La anemia y la parasitemia maternas impiden sustancialmente el transporte de nutrientes a través de la placenta, lo que puede provocar una deprivación de nutrientes para el feto en desarrollo.
Trypanosoma cruzi.
La enfermedad de Chagas es causada por la
infección por T. cruzi, típicamente por un vector artrópodo. La mayoría de las
mujeres en edad reproductiva en áreas endémicas están infectadas y ~ 5%
transmiten el parásito verticalmente211. La mayoría de los lactantes con
infección congénita son asintomáticos, pero pueden desarrollar una enfermedad
incapacitante y potencialmente mortal más adelante en la vida. Las
características de la enfermedad de Chagas congénita sintomática incluyen score Apgar bajo, recién nacidos pequeños para la edad gestacional y
signos de insuficiencia hepática e hidrops212,213.
Diferentes cepas de T. cruzi demuestran un tropismo diferencial por el tejido
placentario, aunque se desconoce la base mecánica de esto. La parasitemia
materna alta se correlaciona con la transmisión congénita214,215. A pesar de que
la patología placentaria demuestra una predilección del parásito por la decidua,
los explantes placentarios ex vivo demostraron que el parásito interactúa con el
STB y causa destrucción y desprendimiento local, probablemente a través de la
apoptosis216,217. Curiosamente, los exosomas derivados de T. cruzi, un marcador
de aumento de la carga parasitaria y enfermedad grave, causan inflamación y daño
histopatológico al STB en explantes placentarios218, lo que sugiere que estos exosomas pueden provocar daño mediado por inflamación y permitir que el parásito
cruce la barrera sincitial.
Los mecanismos parasitarios responsables del daño tisular son un área de investigación en curso, pero se cree que una vez que el tripomastigoto obtiene acceso a la matriz extracelular intervellosa, su degradación promueve el acceso del parásito a la circulación fetal216.
Conclusiones
Las infecciones durante el embarazo tienen
consecuencias sustanciales. La complejidad y las características únicas de la
interfaz materno-fetal han llevado al descubrimiento de múltiples vías de
interacciones huésped-patógeno únicas en este nicho. Sin embargo, los mecanismos
moleculares de la patogenia permanecen en gran parte sin caracterizar, en parte
debido a la complejidad de definir las interacciones que ocurren entre el
patógeno y los huéspedes maternos y / o fetales durante el contexto del
embarazo.
Además, el modelado de la arquitectura
tisular única y la inmunología de la interfaz materno-fetal crea complejidades
adicionales al delinear las estrategias de transmisión vertical microbiana.
Aunque el uso de modelos de ratón ha proporcionado información importante sobre
varios aspectos del embarazo, existen diferencias sustanciales en la
arquitectura placentaria entre humanos y ratones (revisados en las referencias
16, 219) que limitan los correlatos directos de estos hallazgos a los humanos.
Aunque las placentas de primates y cobayas tienen una arquitectura más similar a
la de los humanos, estos modelos pueden ser difíciles de establecer y su falta
de trazabilidad genética limita algunos estudios mecanicistas.
Por último, tanto las muestras clínicas como los tejidos primarios proporcionan modelos basados en humanos para estudiar los mecanismos patógenos o la transmisión vertical de patógenos TORCH; sin embargo, puede haber un acceso limitado a placentas sanas para generar estos modelos. Recientemente, el desarrollo de modelos organoides derivados de células madre de los tipos de células maternas y fetales220,221 en la interfaz materno-fetal ha abierto nuevas y emocionantes vías para modelar esta interfaz. Sin embargo, los modelos organoides carecen de componentes de células inmunes y no recapitulan la diafonía inmunológica que indudablemente altera las defensas antimicrobianas. Los fenotipos de células inmunitarias fetales y maternas en la interfase materno-fetal siguen siendo un área de investigación y caracterización activas. Una mayor comprensión de la contribución de las poblaciones de células inmunitarias enriquecidas en la interfaz será esencial para definir mejor los mecanismos de patogénesis en el embarazo.
Los estudios futuros que aclaren la complejidad de las interacciones entre los tejidos maternos y fetales y cómo estas interacciones son moduladas por patógenos son cruciales para el desarrollo de terapias dirigidas. Además, comprender los mecanismos de la patogénesis microbiana a través de la interfaz materno-fetal tiene implicaciones más amplias en el estudio de la infertilidad, el aborto espontáneo y los trastornos hipertensivos del embarazo y el parto, que pueden tener una etiología común. Aunque la complejidad del embarazo y las brechas actuales en la comprensión de muchos de los aspectos fundamentales de las infecciones en la interfaz materno-fetal representan un desafío, una mayor investigación de este aspecto crucial de la salud humana conducirá a estrategias que podrían mejorar sustancialmente la salud maternoinfantil. .
Referencias
DeSilva, M. et al. Congenital anomalies: case definition and guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine 34, 6015–6026 (2016).
Boyle, B. et al. Estimating global
burden of disease due to congenital anomaly: an analysis of European data.
Arch. Dis. Child. Fetal Neonatal Ed. 103,
F22–F28 (2018).
Christianson, A., Howson, C. & Modell,
B. March of Dimes. Global Report on Birth Defect. The Hidden toll of Dying
and Disabled Children (March of Dimes Birth
Defects Foundation, 2006).
Page, J. M. et al. Stillbirth associated with infection in a diverse U.S. Cohort. Obstet. Gynecol. 134, 1187–1196 (2019).
Lawn, J. E. et al. Stillbirths: rates, risk factors, and acceleration towards 2030. Lancet 387, 587–603 (2016).
McClure, E. M. & Goldenberg, R. L. Infection and stillbirth. Semin. Fetal Neonatal Med. 14, 182–189 (2009).
Fouks, Y., Many, A., Shulman, Y., Bak, S. & Shinar, S. The contribution of an infectious workup in understanding stillbirth. Am. J. Perinatol. 38, 377–382 (2021).
Fiumara, N. J. A legacy of syphilis. Arch. Dermatol. 92, 676–678 (1965).
Crane, J. et al. Parvovirus B19 infection in pregnancy. J. Obstet. Gynaecol. Can. 36, 1107–1116 (2014).
Romero, R., Dey, S. K. & Fisher, S. J. Preterm labor: one syndrome, many causes. Science 345, 760–765 (2014).
Venkatesh, K. K. et al. Association of chorioamnionitis and its duration with neonatal morbidity and mortality. J. Perinatol. 39, 673–682 (2019).
Stoll, B. J. et al. Changes in pathogens causing early- onset sepsis in very- low-birth- weight infants. N. Engl. J. Med. 347, 240–247 (2002).
Oh, J. W., Park, C. W., Moon, K. C.,
Park, J. S. & Jun, J. K. The relationship among the progression of
inflammation in umbilical cord, fetal inflammatory
response, early- onset neonatal sepsis, and chorioamnionitis. PLoS ONE 14,
e0225328 (2019).
Stoll, B. J. et al. Early- onset
neonatal sepsis 2015 to 2017, the rise of Escherichia coli, and the need for
novel prevention strategies. JAMA Pediatr. 174,
e200593 (2020).
Nahmias, A. J., Walls, K. W., Stewart, J. A., Herrmann, K. L. & Flynt, W. J. The ToRCH complexperinatal infections associated with toxoplasma and rubella, cytomegol- and herpes simplex viruses. Pediatr. Res. 5, 405–406 (1971).
Ander, S. E., Diamond, M. S. & Coyne, C. B. Immune responses at the maternal- fetal interface. Sci. Immunol. 4, eaat6114 (2019).
Maltepe, E., Bakardjiev, A. I. &
Fisher, S. J. The placenta: transcriptional, epigenetic, and physiological
integration during development. J. Clin. Invest. 120,
1016–1025 (2010).
Knöfler, M. et al. Human placenta and trophoblast development: key molecular mechanisms and model systems. Cell. Mol. Life Sci. 76, 405–406 (2019).
Thomas, J. R. et al. Phenotypic and functional characterization of first- trimester human placental macrophages, Hofbauer cells. J. Exp. Med. 218, e20200891 (2020).
Ellery, P. M., Cindrova- Davies, T.,
Jauniaux, E., Ferguson- Smith, A. C. & Burton, G. J. Evidence for
transcriptional activity in the syncytiotrophoblast
of the human placenta. Placenta 30, 329–334 (2009).
Guttman, J. A. & Finlay, B. B. Tight junctions as targets of infectious agents. Biochim. Biophys. Acta 1788, 832–841 (2009).
Zeldovich, V. B. et al. Placental syncytium forms a biophysical barrier against pathogen invasion. PLoS Pathog. 9, 1–10 (2013).
Ander, S. E. et al. Human placental syncytiotrophoblasts restrict Toxoplasma gondii attachment and replication and respond to infection by producing immunomodulatory chemokines. mBio 9, e01678-17 (2018).
Robbins, J. R., Skrzypczynska, K. M.,
Zeldovich, V. B., Kapidzic, M. & Bakardjiev, A. I. Placental
syncytiotrophoblast constitutes a major barrier
to vertical transmission of Listeria monocytogenes. PLoS Pathog. 6, e1000732
(2010).
Delorme- Axford, E. et al. Human placental trophoblasts confer viral resistance to recipient cells. Proc. Natl Acad. Sci. USA 110, 12048–12053 (2013).
Delorme- Axford, E., Sadovsky, Y. & Coyne, C. B. Lipid raft- and src family kinase- dependent entry of Coxsackievirus B into human placental trophoblasts. J. Virol. 87, 8569–8581 (2013).
Stein, K. R. et al. CD46 facilitates entry and dissemination of human cytomegalovirus. Nat. Commun. 10, 2699 (2019).
Hemmings, D. G., Kilani, R., Nykiforuk, C., Preiksaitis, J. & Guilbert, L. J. Permissive cytomegalovirus infection of primary villous term and first trimester trophoblasts. J. Virol. 72, 4970–4979 (1998).
Vento- Tormo, R. et al. Single- cell reconstruction of the early maternal–fetal interface in humans. Nature 563, 347–353 (2018).
Pique- Regi, R. et al. Single cell transcriptional signatures of the human placenta in term and preterm parturition. eLife 8, e52004 (2019).
Rinaldi, S. F., Makieva, S., Saunders, P. T., Rossi, A. G. & Norman, J. E. Immune cell and transcriptomic analysis of the human decidua in term and preterm parturition. Mol. Hum. Reprod. 23, 708–724 (2017).
Hamilton, S. et al. Macrophages Infiltrate the human and rat decidua during term and preterm labor: evidence that decidual inflammation precedes labor1. Biol. Reprod. 86, 39 (2012).
Kwan, M. et al. Dynamic changes in maternal decidual leukocyte populations from first to second trimester gestation. Placenta 35, 1027–1034 (2014).
Smith, S. D., Dunk, C. E., Aplin, J. D., Harris, L. K. & Jones, R. L. Evidence for immune cell involvement in decidual spiral arteriole remodeling in early human pregnancy. Am. J. Pathol. 174, 1959–1971 (2009).
Harris, L. K., Benagiano, M., D’Elios,
M. M., Brosens, I. & Benagiano, G. Placental bed research: II. Functional
and immunological investigations of the
placental bed. Am. J. Obstet. Gynecol. 221, 457–469 (2019).
Pereira, L. & Maidji, E. Cytomegalovirus infection in the human placenta: maternal immunity and developmentally regulated receptors on trophoblasts converge. Curr. Top. Microbiol. Immunol. 325, 383–395 (2008).
Weisblum, Y. et al. Zika virus infects early- and midgestation human maternal decidual tissues, inducing distinct innate tissue responses in the maternal- fetal interface. J. Virol. 91, e01905–e01916 (2017).
Rizzuto, G., Tagliani, E., Manandhar, P., Erlebacher, A. & Bakardjiev, A. I. Limited colonization undermined by inadequate early immune responses defines the dynamics of decidual listeriosis. Infect. Immun. 85, e00153–17 (2017).
Ning, F., Liu, H. & Lash, G. E. The role of decidual macrophages during normal and pathological pregnancy. Am. J. Reprod. Immunol. 75, 298–309 (2016).
Jiang, X., Du, M. R., Li, M. & Wang, H. Three macrophage subsets are identified in the uterus during early human pregnancy. Cell. Mol. Immunol. 15, 1027–1037 (2018).
Crespo, Â. C. et al. Decidual NK cells transfer granulysin to selectively kill bacteria in trophoblasts. Cell 182, 1125–1139.e18 (2020).
Rogers, L. M. et al. Decidual stromal cell- derived PGE2 regulates macrophage responses to microbial threat. Am. J. Reprod. Immunol. 80, e13032 (2018).
Croxatto, D. et al. Stromal cells from human decidua exert a strong inhibitory effect on NK cell function and dendritic cell differentiation. PLoS ONE 9, e89006 (2014).
Castro- Leyva, V. et al. Decidualization mediated by steroid hormones modulates the innate immunity in response to group B streptococcal infection in vitro. Gynecol. Obstet. Invest. 82, 592–600 (2017).
Xu, X. et al. Monocyte chemoattractant protein-1 secreted by decidual stromal cells inhibits NK cells cytotoxicity by up- regulating expression of SOCS3. PLoS ONE 7, e41869 (2012).
Guzeloglu- Kayisli, O. et al. Zika
virus–infected decidual cells elicit a gestational age–dependent innate
immune response and exaggerate trophoblast
zika permissiveness: implication for vertical transmission. J. Immunol. 205,
3083–3094 (2020).
Tabata, T., Petitt, M., Fang- Hoover, J. & Pereira, L. Survey of cellular immune responses to human cytomegalovirus infection in the microenvironment of the uterine–placental interface. Med. Microbiol. Immunol. 208, 475–485 (2019).
Bortolotti, D. et al. Human herpes simplex 1 virus infection of endometrial decidual tissue- derived MSC alters HLA- G expression and immunosuppressive functions. Hum. Immunol. 79, 800–808 (2018).
Deshmukh, H. & Way, S. S. Immunological basis for recurrent fetal loss and pregnancy complications. Annu. Rev. Pathol. Mech. Dis. 14, 185–210 (2019).
Reyes, L. & Golos, T. G. Hofbauer cells: their role in healthy and complicated pregnancy. Front. Immunol. 9, 2628 (2018).
Bayer, A. et al. Type III interferons produced by human placental trophoblasts confer protection against Zika virus infection. Cell Host Microbe 19, 705–712 (2016).
Corry, J., Arora, N., Good, C. A., Sadovsky, Y. & Coyne, C. B. Organotypic models of type III interferonmediated protection from Zika virus infections at the maternal- fetal interface. Proc. Natl Acad. Sci. USA 114, 9433–9438 (2017).
Jagger, B. W. et al. Gestational stage and IFN- λ signaling regulate ZIKV infection in utero. Cell Host Microbe 22, 366–376.e3 (2017).
Chen, J. et al. Outcomes of congenital Zika disease depend on timing of infection and maternal- fetal interferon action. Cell Rep. 21, 1588–1599 (2017).
Bayer, A. et al. Chromosome 19 microRNAs exert antiviral activity independent from type III interferon signaling. Placenta 61, 33–38 (2018).
Bayer, A. et al. Human trophoblasts confer resistance to viruses implicated in perinatal infection. Am. J. Obstet. Gynecol. 212, 71.e1–71.e8 (2015).
Dumont, T. M. F. et al. The expression level of C19MC miRNAs in early pregnancy and in response to viral infection. Placenta 53, 23–29 (2017).
Yockey, L. J. et al. Type I interferons instigate fetal demise after Zika virus infection. Sci. Immunol. 3, eaao1680 (2018).
Buchrieser, J. et al. IFITM proteins inhibit placental syncytiotrophoblast formation and promote fetal demise. Science 365, 176–180 (2019).
Zani, A. et al. Interferon- induced transmembrane proteins inhibit cell fusion mediated by trophoblast syncytins. J. Biol. Chem. 294, 19844–19851 (2019).
Robbins, J. R., Zeldovich, V. B., Poukchanski, A., Boothroyd, J. C. & Bakardjiev, A. I. Tissue barriers of the human placenta to infection with Toxoplasma gondii. Infect. Immun. 80, 418–428 (2012).
Megli, C., Morosky, S., Rajasundaram, D. & Coyne, C. B. Inflammasome signaling in human placental trophoblasts regulates immune defense against Listeria monocytogenes infection. J. Exp. Med. 218, e20200649 (2021).
Reis, A. S. et al. Inflammasome activation and IL-1 signaling during placental malaria induce poor pregnancy outcomes. Sci. Adv. 6, eaax6346 (2020).
Thomas, J. et al. Outbreak of listeriosis in South Africa associated with processed meat. N. Engl. J. Med. 382, 632–643 (2020).
Chan, B. T., Hohmann, E., Barshak, M. B. & Pukkila- Worley, R. Treatment of listeriosis in first trimester of pregnancy. Emerg. Infect. Dis. 19, 839–841 (2013).
Chan, L. M., Lin, H. H. & Hsiao, S. M. Successful treatment of maternal Listeria monocytogenes bacteremia in the first trimester of pregnancy: a case report and literature review. Taiwan. J. Obstet. Gynecol. 57, 462–463 (2018).
Al- Tawfiq, J. A. Listeria monocytogenes bacteremia in a twin pregnancy with differential outcome: fetus papyraceus and a full- term delivery. J. Microbiol. Immunol. Infect. 41, 433–436 (2008).
Mylonakis, E., Paliou, M., Hohmann, E.
L., Calderwood, S. B. & Wing, E. J. Listeriosis during pregnancy: a case
series and review of 222 cases.
Medicine 81, 260–269 (2002).
Phelps, C. C. et al. Relative roles of Listeriolysin O, InlA, and InlB in Listeria monocytogenes uptake by host cells. Infect. Immun. 86, e00555-18 (2018).
Lecuit, M. et al. Targeting and crossing of the human maternofetal barrier by Listeria monocytogenes: role of internalin interaction with trophoblast E- cadherin. Proc. Natl Acad. Sci. USA 101, 6152–6157 (2004).
Holch, A., Ingmer, H., Licht, T. R. & Gram, L. Listeria monocytogenes strains encoding premature stop codons in inlA invade mice and guinea pig fetuses in orally dosed dams. J. Med. Microbiol. 62, 1799–1806 (2013).
Disson, O. et al. Conjugated action of two speciesspecific invasion proteins for fetoplacental listeriosis. Nature 455, 1114–1118 (2008).
Morrison, H. A., Lowe, D., Robbins, J. R. & Bakardjiev, A. I. In vivo virulence characterization of pregnancy- associated Listeria monocytogenes infections. Infect. Immun. 86, e00397-18 (2018).
Bakardjiev, A. I., Theriot, J. A. & Portnoy, D. A. Listeria monocytogenes traffics from maternal organs to the placenta and back. PLoS Pathog. 2, 623–631 (2006).
Le Monnier, A. et al. ActA is required for crossing of the fetoplacental barrier by Listeria monocytogenes. Infect. Immun. 75, 950–957 (2007).
Faralla, C. et al. Listeria monocytogenes InlP interacts with afadin and facilitates basement membrane crossing. PLoS Pathog. 14, e1007094 (2018).
Parkash, V. et al. Immunohistochemical detection of Listeria antigens in the placenta in perinatal listeriosis. Int. J. Gynecol. Pathol. 17, 343–350 (1998).
Wolfe, B. et al. Sequelae of fetal infection in a nonhuman primate model of listeriosis. Front. Microbiol. 10, 2021 (2019).
Radolf, J. D. & Kumar, S. The Treponema pallidum outer membrane. Curr. Topics Microbiol. Immunol. 415, 1–38 (2018).
Gomez, G. B. et al. Untreated maternal syphilis and adverse outcomes of pregnancy: a systematic review and meta- analysis. Bull. World Health Organ. 91, 217–226 (2013).
Korenromp, E. L. et al. Global burden of maternal and congenital syphilis and associated adverse birth outcomes — estimates for 2016 and progress since 2012. PLoS ONE 14, e0211720 (2019).
Newman, L. et al. Global estimates of syphilis in pregnancy and associated adverse outcomes: analysis of multinational antenatal surveillance data. PLoS Med. 10, e1001396 (2013).
Peeling, R. W. & Hook, E. W. The pathogenesis of syphilis: the Great Mimicker, revisited. J. Pathol. 208, 224–232 (2006).
Wicher, V. & Wicher, K. Pathogenesis of maternal-fetal syphilis revisited. Clin. Infect. Dis. 33, 354–363 (2001).
Rac, M. W. F. et al. Progression of ultrasound findings of fetal syphilis after maternal treatment. Am. J. Obstet. Gynecol. 211, 426.e1–426.e6 (2014).
Kimball, A. et al. Missed opportunities for prevention of congenital syphilis — United States, 2018. MMWR 69, 661–665 (2020).
Balaji, G. & Kalaivani, S. Observance of Kassowitz law — late congenital syphilis: palatal perforation and saddle nose deformity as presenting features. Indian J. Sex. Transm. Dis. 34, 35–37 (2013).
Kassowitz, M. Die Vererbung der Syphilis (Kessinger Publishing, 1876).
Dhanaselvi, H. & Kalaivani, S. Untreated late latent syphilis of both spouses with observation of Kassowitz law: adverse pregnancy outcomes in the postpenicillin era. Indian. J. Dermatol. 62, 221–222 (2017).
Wicher, V., Baughn, R. E. & Wicher, K. Congenital and neonatal syphilis in guinea- pigs show a different pattern of immune response. Immunology 82, 404–409 (1994).
Wicher, K., Abbruscato, F., Wicher, V., Baughn, R. & Noordhoek, G. T. Target organs of infection in guinea pigs with acquired or congenital syphilis. Infect. Immun. 64, 3174–3179 (1996).
Sheffield, J. S. et al. Placental histopathology of congenital syphilis. Obstet. Gynecol. 100, 126–133 (2002).
Genest, D. R. et al. Diagnosis of congenital syphilis from placental examination: comparison of histopathology, steiner stain, and polymerase chain reaction for Treponema pallidum DNA. Hum. Pathol. 27, 366–372 (1996).
Myles, T. D., Elam, G., Park- Hwang, E. & Nguyen, T. The Jarisch–Herxheimer reaction and fetal monitoring changes in pregnant women treated for syphilis. Obstet. Gynecol. 92, 859–864 (1998).
Romero, R. et al. The role of inflammation and infection in preterm birth. Semin. Reprod. Med. 25, 21–39 (2007).
Nagy, I., Pap, K., Dicső, F. & Arany, I. Chorioamnionitis is still the main cause of preterm birth. Eur. J. Obstet. Gynecol. Reprod. Biol. 206, e84 (2016).
Mendz, G. L., Kaakoush, N. O. & Quinlivan, J. A. Bacterial aetiological agents of intra- amniotic infections and preterm birth in pregnant women. Front. Cell. Infect. Microbiol. 3, 58 (2013).
Romero, R. et al. Sterile and microbial- associated intra- amniotic inflammation in preterm prelabor rupture of membranes. J. Matern. Fetal. Neonatal Med. 28, 1394–1409 (2015).
Schrag, S. J. et al. A population- based comparison of strategies to prevent early- onset group B streptococcal disease in neonates. N. Engl. J. Med. 347, 233– 239 (2002).
Ayala, O. D. et al. Raman microspectroscopy differentiates perinatal pathogens on ex vivo infected human fetal membrane tissues. J. Biophotonics 12, e201800449 (2019).
Verani, J. R., McGee, L., Schrag, S. J. & Division of Bacterial Diseases, National Center for Immunization and Respiratory Diseases, Centers for Disease Control and Prevention (CDC). Prevention of perinatal group B streptococcal disease — revised guidelines from CDC, 2010. MMWR Recomm. Rep. 59, 1–36 (2010).
Doster, R. S., Sutton, J. A., Rogers,
L. M., Aronoff, D. M. & Gaddy, J. A. Streptococcus agalactiae induces
placental macrophages to release extracellular traps
loaded with tissue remodeling enzymes via an oxidative burst- dependent
mechanism. mBio 9, e02084-18 (2018).
Kothary, V. et al. Group B Streptococcus induces neutrophil recruitment to gestational tissues and elaboration of extracellular traps and nutritional immunity. Front. Cell. Infect. Microbiol. 7, 19 (2017).
Armistead, B., Oler, E., Adams Waldorf,
K. & Rajagopal, L. The double life of group B Streptococcus:
asymptomatic colonizer and potent pathogen. J. Mol.
Biol. 431, 2914–2931 (2019).
Vornhagen, J., Adams Waldorf, K. M. &
Rajagopal, L. Perinatal group B streptococcal infections: virulence factors,
immunity, and prevention strategies. Trends
Microbiol. 25, 919–931 (2017).
Galask, R. P., Varner, M. W., Rosemarie Petzold, C. & Wilbur, S. L. Bacterial attachment to the chorioamniotic membranes. Am. J. Obstet. Gynecol. 148, 915–928 (1984).
Wilkie, G. L. et al. Microbiology and antibiotic resistance in peripartum bacteremia. Obstet. Gynecol. 133, 269–275 (2019).
Sáez-López, E. et al. Vaginal versus obstetric infection Escherichia coli isolates among pregnant women: antimicrobial resistance and genetic virulence profile. PLoS ONE 11, e0146531 (2016).
Pathirana, J. et al. Prevalence of congenital cytomegalovirus infection and associated risk of in- utero HIV acquisition in a high HIV prevalence setting, South Africa. Clin. Infect. Dis. 69, 1789–1796 (2019).
Fowler, K. B. et al. The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. N. Engl. J. Med. 326, 663–667 (1992).
Boppana, S. B., Rivera, L. B., Fowler, K. B., Mach, M. & Britt, W. J. Intrauterine transmission of cytomegalovirus to infants of women with preconceptional immunity. N. Engl. J. Med. 344, 1366–1371 (2001).
Enders, G., Daiminger, A., Bäder, U., Exler, S. & Enders, M. Intrauterine transmission and clinical outcome of 248 pregnancies with primary cytomegalovirus infection in relation to gestational age. J. Clin. Virol. 52, 244–246 (2011).
Uenaka, M. et al. Histopathological analysis of placentas with congenital cytomegalovirus infection. Placenta 75, 62–67 (2019).
Satosar, A., Ramirez, N. C., Bartholomew, D., Davis, J. & Nuovo, G. J. Histologic correlates of viral and bacterial infection of the placenta associated with severe morbidity and mortality in the newborn. Hum. Pathol. 35, 536–545 (2004).
Njue, A. et al. The role of congenital cytomegalovirus infection in adverse birth outcomes: a review of the potential mechanisms. Viruses 13, 20 (2020).
Schleiss, M. R. Congenital cytomegalovirus infection: molecular mechanisms mediating viral pathogenesis. Infect. Disord. Drug Targets 11, 449–465 (2011).
Britt, W. J. Human cytomegalovirus infection in women with preexisting immunity: sources of infection and mechanisms of infection in the presence of antiviral immunity. J. Infect. Dis. 221, S1–S8 (2020).
Schleiss, M. R. & McVoy, M. A. Guinea pig cytomegalovirus: a model for the prevention and treatment of maternal–fetal cytomegalovirus transmission. Future Virol. 5, 207–217 (2010).
Weisblum, Y. et al. Modeling of human
cytomegalovirus maternal–fetal transmission in a novel decidual organ
culture. J. Virol. 85,
13204–13213 (2011).
Aronoff, D. M., Correa, H., Rogers, L. M., Arav- Boger, R. & Alcendor, D. J. Placental pericytes and cytomegalovirus infectivity: implications for HCMV placental pathology and congenital disease. Am. J. Reprod. Immunol. https://doi.org/10.1111/aji.12728 (2017).
Burgos, J. S. et al. Hematogenous vertical transmission of herpes simplex virus type 1 in mice. J. Virol. 80, 2823–2831 (2006).
Koi, H. et al. Syncytiotrophoblast is a barrier to maternal–fetal transmission of herpes simplex virus. Biol. Reprod. 67, 1572–1579 (2002).
Finger- Jardim, F. et al. Herpes simplex virus: prevalence in placental tissue and incidence in neonatal cord blood samples. J. Med. Virol. 86, 519–524 (2014).
Finger- Jardim, F. et al. Prevalence of herpes simplex virus types 1 and 2 at maternal and fetal sides of the placenta in asymptomatic pregnant women. Am. J. Reprod. Immunol. 78, e12689 (2017).
Gay, N. J. et al. Age specific antibody prevalence to parvovirus B19: how many women are infected in pregnancy? Commun. Dis. Rep. CDR Rev. 4, R104–7 (1994).
Li, J. J., Henwood, T., Van Hal, S. & Charlton, A. Parvovirus infection: an immunohistochemical study using fetal and placental tissue. Pediatr. Dev. Pathol. 18, 30–39 (2015).
Jordan, J. A. & Deloia, J. A. Globoside expression within the human placenta. Placenta 20, 103–108 (1999).
Wegner, C. C. & Jordan, J. A. Human parvovirus B19 VP2 empty capsids bind to human villous trophoblast cells in vitro via the globoside receptor. Infect. Dis. Obstet. Gynecol. 12, 69–78 (2004).
Moffatt, S., Yaegashi, N., Tada, K.,
Tanaka, N. & Sugamura, K. Human parvovirus B19 nonstructural (NS1) protein
induces apoptosis in erythroid lineage
cells. J. Virol. 72, 3018–3028 (1998).
Jordan, J. A. & Butchko, A. R. Apoptotic activity in villous trophoblast cells during B19 infection correlates with clinical outcome: assessment by the caspase- related M30 cytodeath antibody. Placenta 23, 547–553 (2002).
Dickinson, J. E., Keil, A. D. & Charles, A. K. Discordant fetal infection for parvovirus B19 in a dichorionic twin pregnancy. Twin Res. Hum. Genet. 9, 456–459 (2006).
Schiesser, M., Sergi, C., Enders, M., Maul, H. & Schnitzler, P. Discordant outcomes in a case of parvovirus B19 transmission into both dichorionic twins. Twin Res. Hum. Genet. 12, 175–179 (2009). www.nature.com/nrmicro
Bernstein, H. B. & Wegman, A. D. HIV infection: antepartum treatment and management. Clin. Obstet. Gynecol. 61, 122–136 (2018).
Maury, W., Potts, B. J. & Rabson, A. B. HIV-1 infection of first- trimester and term human placental tissue: a possible mode of maternal- fetal transmission. J. Infect. Dis. 160, 583–588 (1989).
Amirhessami- Aghili, N. & Spector, S. A. Human immunodeficiency virus type 1 infection of human placenta: potential route for fetal infection. Dis. Markers 9, 348 (1991).
Mattern, C. F. T. et al. Localization of human immunodeficiency virus core antigen in term human placentas. Pediatrics 89, 207–209 (1992).
Peuchmaur, M. et al. HIV proteins absent from placentas of 75 HIV-1-positive women studied by immunohistochemistry. AIDS 5, 741–745 (1991).
Backe, E. et al. Demonstration of HIV-1 infected cells in human placenta by in situ hybridisation and immunostaining. J. Clin. Pathol. 45, 871–874 (1992).
Townsend, C. L. et al. Low rates of mother- to-child transmission of HIV following effective pregnancy interventions in the United Kingdom and Ireland, 2000– 2006. AIDS 22, 973–981 (2008).
Koay, W. L. A. et al. Prevention of perinatal HIV transmission in an area of high HIV prevalence in the United States. J. Pediatr. 228, 101–109 (2021).
Johnson, E. L. & Chakraborty, R. HIV-1 at the placenta: immune correlates of protection and infection. Curr. Opin. Infect. Dis. 29, 248–255 (2016).
Mlakar, J. et al. Zika virus associated with microcephaly. N. Engl. J. Med. 374, 951–958 (2016).
Brady, O. J. et al. The association between Zika virus infection and microcephaly in Brazil 2015–2017: an observational analysis of over 4 million births. PLoS Med. 16, e1002755 (2019).
Hoen, B. et al. Pregnancy outcomes after ZIKV infection in French territories in the Americas. N. Engl. J. Med. 378, 985–994 (2018).
van der Linden, V. et al. Description of 13 infants born during October 2015–January 2016 with congenital Zika virus infection without microcephaly at birth — Brazil. MMWR Morb. Mortal. Wkly Rep. 65, 1343–1348 (2016).
Sobhani, N. C. et al. Discordant Zika virus findings in twin pregnancies complicated by antenatal Zika virus exposure: a prospective cohort. J. Infect. Dis. 221, 1838–1845 (2020).
Amaral, M. S. et al. Differential gene expression elicited by ZIKV infection in trophoblasts from congenital Zika syndrome discordant twins. PLoS Negl. Trop. Dis. 14, e0008424 (2020).
Hastings, A. K. et al. TAM receptors are not required for Zika virus infection in mice. Cell Rep. 19, 558–568 (2017).
Hermanns, K. et al. Zika virus infection in human placental tissue explants is enhanced in the presence of dengue virus antibodies in- vitro. Emerg. Microbes Infect. 7, 198 (2018).
Zimmerman, M. G. et al. Cross- reactive dengue virus antibodies augment Zika virus infection of human placental macrophages. Cell Host Microbe 24, 731–742.e6 (2018).
Rodriguez- Barraquer, I. et al. Impact of preexisting dengue immunity on Zika virus emergence in a dengue endemic region. Science 363, 607–610 (2019).
Pedroso, C. et al. Cross- protection of dengue virus infection against congenital Zika syndrome, northeastern Brazil. Emerg. Infect. Dis. 25, 1485–1493 (2019).
Pantoja, P. et al. Zika virus pathogenesis in rhesus macaques is unaffected by pre- existing immunity to dengue virus. Nat. Commun. 8, 15674 (2017).
Breitbach, M. E. et al. Primary infection with dengue or Zika virus does not affect the severity of heterologous secondary infection in macaques. PLoS Pathog. 15, e1007766 (2019).
Katzelnick, L. C. et al. Zika virus infection enhances future risk of severe dengue disease. Science 369, 1123–1128 (2020).
Dudley, D. M. et al. Using macaques to address critical questions in Zika virus research. Annu. Rev. Virol. 6, 481–500 (2019).
Koenig, M. R. et al. Quantitative definition of neurobehavior, vision, hearing and brain volumes in macaques congenitally exposed to Zika virus. PLoS ONE 15, e0235877 (2020).
Bebell, L. M., Oduyebo, T. & Riley, L. E. Ebola virus disease and pregnancy: a review of the current knowledge of Ebola virus pathogenesis, maternal, and neonatal outcomes. Birth Defects Res. 109, 353–362 (2017).
Muehlenbachs, A. et al. Ebola virus disease in pregnancy: clinical, histopathologic and immunohistochemical findings. J. Infect. Dis. 215, 64–69 (2015).
Oduyebo, T. et al. A pregnant patient with ebola virus disease. Obstet. Gynecol. 126, 1273–1275 (2015).
Baudin, M. et al. Association of Rift Valley fever virus infection with miscarriage in Sudanese women: a cross- sectional study. Lancet Glob. Heal. 4, e864–e871 (2016).
Arishi, H. M., Aqeel, A. Y. & Al Hazmi, M. M. Vertical transmission of fatal Rift Valley fever in a newborn. Ann. Trop. Paediatr. 26, 251–253 (2006).
Oymans, J., Wichgers Schreur, P. J., van Keulen, L., Kant, J. & Kortekaas, J. Rift valley fever virus targets the maternal–foetal interface in ovine and human placentas. PLoS Negl. Trop. Dis. 14, 1–18 (2020).
McMillen, C. M. et al. Rift Valley fever virus induces fetal demise in Sprague–Dawley rats through direct placental infection. Sci. Adv. 4, eaau9812 (2018).
O’Leary, D. R. et al. Birth outcomes following west nile virus infection of pregnant women in the United States: 2003–2004. Pediatrics 117, e537–e545 (2006).
Platt, D. J. et al. Zika virus- related neurotropic flaviviruses infect human placental explants and cause fetal demise in mice. Sci. Transl. Med. 10, eaao7090 (2018).
Julander, J. G. et al. West Nile virus infection of the placenta. Virology 347, 175–182 (2006).
Chen, H. et al. Clinical characteristics and intrauterine vertical transmission potential of COVID-19 infection in nine pregnant women: a retrospective review of medical records. Lancet 395, 809–815 (2020).
Pique- Regi, R. et al. Does the human placenta express the canonical cell entry mediators for SARS- CoV-2? eLife 9, e58716 (2020).
Li, Y. et al. Lack of vertical transmission of severe acute respiratory syndrome coronavirus 2, China. Emerg. Infect. Dis. 26, 1335–1336 (2020).
Egloff, C., Vauloup- Fellous, C., Picone, O., Mandelbrot, L. & Roques, P. Evidence and possible mechanisms of rare maternal- fetal transmission of SARS- CoV- 2. J. Clin. Virol. 128, 104447 (2020).
WAPM working group on COVID-19. . Maternal and perinatal outcomes of pregnant women with SARS- COV-2 infection. Ultrasound Obstet. Gynecol. 57, 232–241 (2021).
Flaherman, V. J. et al. Infant outcomes following maternal infection with SARS- CoV-2: first report from the PRIORITY study. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa1411 (2020).
Torgerson, P. R. & Mastroiacovo, P. La charge mondiale de la toxoplasmose: une étude systématique. Bull. World Health Organ. 91, 501–508 (2013).
Galanakis, E. et al. Outcome of toxoplasmosis acquired during pregnancy following treatment in both pregnancy and early infancy. Fetal Diagn. Ther. 22, 444–448 (2007).
Berrébi, A. et al. Long- term outcome of children with congenital toxoplasmosis. Am. J. Obstet. Gynecol. 203, 552.e1–552.e6 (2010).
Dunn, D. et al. Mother- to-child transmission of toxoplasmosis: risk estimates for clinical counselling. Lancet 353, 1829–1833 (1999).
Mendez, O. A. & Koshy, A. A. Toxoplasma gondii: entry, association, and physiological influence on the central nervous system. PLoS Pathog. 13, e1006351 (2017).
Bisio, H. & Soldati- Favre, D. Signaling cascades governing entry into and exit from host cells by Toxoplasma gondii. Annu. Rev. Microbiol. 73, 579–599 (2019).
English, E. D., Adomako- Ankomah, Y. & Boyle, J. P. Secreted effectors in Toxoplasma gondii and related species: determinants of host range and pathogenesis? Parasite Immunol. 37, 127–140 (2015).
Hunter, C. A. & Sibley, L. D. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat. Rev. Microbiol. 10, 766–778 (2012).
Rudzki, E. N. et al. Toxoplasma gondii GRA28 is required for specific induction of the regulatory chemokine CCL22 in human and mouse cells. Preprint at bioRxiv https://doi.org/10.1101/2020.10.14.335802 (2020).
Luxemburger, C. et al. Effects of malaria during pregnancy on infant mortality in an area of low malaria transmission. Am. J. Epidemiol. 154, 459–465 (1993).
Luxemburger, C. et al. The epidemiology of severe malaria in an area of low transmission in Thailand. Trans. R. Soc. Trop. Med. Hyg. 91, 256–262 (1997). 185.
Guyatt, H. L. & Snow, R. W. Malaria in pregnancy as an indirect cause of infant mortality in sub- Saharan Africa. Trans. R. Soc. Trop. Med. Hyg. 95, 569–576 (2001).
Desai, M. et al. Epidemiology and burden of malaria in pregnancy. Lancet Infect. Dis. 7, 93–104 (2007).
Tobian, A. A. et al. Frequent umbilical cord- blood and maternal- blood infections with Plasmodium falciparum, P. malariae, and P. ovale in Kenya. J. Infect. Dis. 182, 558–563 (2000).
Ouédraogo, A. et al. Transplacental transmission of plasmodium falciparum in a highly malaria endemic area of Burkina Faso. J. Trop. Med. 2012, 109705 (2012).
Beeson, J. G., Amin, N., Kanjala, M. & Rogerson, S. J. Selective accumulation of mature asexual stages of Plasmodium falciparum- infected erythrocytes in the placenta. Infect. Immun. 70, 5412–5415 (2002).
Muehlenbachs, A. et al. A novel histological grading scheme for placental malaria applied in areas of high and low malaria transmission. J. Infect. Dis. 202, 1608–1616 (2010).
Okoko, B. J. et al. The influence of placental malaria infection and maternal hypergammaglobulinemia on transplacental transfer of antibodies and IgG subclasses in a rural west African population. J. Infect. Dis. 184, 627–632 (2001).
Owens, S. et al. Placental malaria and immunity to infant measles. Arch. Dis. Child. 91, 507–508 (2006).
Frech, C. & Chen, N. Variant surface antigens of malaria parasites: functional and evolutionary insights from comparative gene family classification and analysis. BMC Genomics 14, 427 (2013).
Beeson, J. G. & Duffy, P. E. The immunology and pathogenesis of malaria during pregnancy. Curr. Top. Microbiol. Immunol. 297, 187–227 (2005).
Salanti, A. et al. Selective upregulation of a single distinctly structured var gene in chondroitin sulphate A- adhering Plasmodium falciparum involved in pregnancy- associated malaria. Mol. Microbiol. 49, 179–191 (2003).
Tuikue Ndam, N. G. et al. High level of var2csa transcription by Plasmodium falciparum isolated from the placenta. J. Infect. Dis. 192, 331–335 (2005).
Magistrado, P. et al. VAR2CSA
expression on the surface of placenta- derived Plasmodium falciparuminfected
erythrocytes. J. Infect. Dis. 198, 1071–1074
(2008).
Chotivanich, K. et al. Plasmodium vivax adherence to placental glycosaminoglycans. PLoS ONE 7, e34509 (2012).
Fried, M. & Duffy, P. E. Adherence of Plasmodium falciparum to chondroitin sulfate A in the human placenta. Science 272, 1502–1504 (1996).
Fried, M., Domingo, G. J., Gowda, C. D., Mutabingwa, T. K. & Duffy, P. E. Plasmodium falciparum: chondroitin sulfate A is the major receptor for adhesion of parasitized erythrocytes in the placenta. Exp. Parasitol. 113, 36–42 (2006).
O’Neil- Dunne, I. et al. Gravidity-
dependent production of antibodies that inhibit binding of Plasmodium
falciparum- infected erythrocytes to placental
chondroitin sulfate proteoglycan during pregnancy. Infect. Immun. 69,
7487–7492 (2001).
Ricke, C. H. et al. Plasma antibodies from malaria- exposed pregnant women recognize variant surface antigens on plasmodium falciparum- infected erythrocytes in a parity- dependent manner and block parasite adhesion to chondroitin sulfate A. J. Immunol. 165, 3309–3316 (2000).
Oleinikov, A. V. et al. Effects of sex, parity, and sequence variation on seroreactivity to candidate pregnancy malaria vaccine antigens. J. Infect. Dis. 196, 155–164 (2007).
Duffy, P. E. & Fried, M. Antibodies
that inhibit plasmodium falciparum adhesion to chondroitin sulfate A are
associated with increased birth weight
and the gestational age of newborns. Infect. Immun. 71, 6620–6623 (2003).
Fried, M. & Duffy, P. E. Malaria during pregnancy. Cold Spring Harb. Perspect. Med. 7, a025551 (2017).
Staalsoe, T. et al. Variant surface antigen- specific IgG and protection against clinical consequences of pregnancy- associated Plasmodium falciparum malaria. Lancet 363, 283–289 (2004).
Rogerson, S. J. et al. Placental
monocyte infiltrates in response to Plasmodium falciparum malaria infection
and their association with adverse pregnancy
outcomes. Am. J. Trop. Med. Hyg. 68, 115–119 (2003).
Moormann, A. M. et al. Malaria and pregnancy: placental cytokine expression and its relationship to intrauterine growth retardation. J. Infect. Dis. 180, 1987–1993 (1999).
Lima, F. A. et al. Plasmodium falciparum infection dysregulates placental autophagy. PLoS ONE 14, e0226117 (2019).
Dimasuay, K. G. et al. Impaired placental autophagy in placental malaria. PLoS ONE 12, e0187291 (2017).
Rios, L., Campos, E. E., Menon, R., Zago, M. P. & Garg, N. J. Epidemiology and pathogenesis of maternal–fetal transmission of Trypanosoma cruzi and a case for vaccine development against congenital Chagas disease. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165591 (2020).
Torrico, F. et al. Maternal trypanosoma cruzi infection, pregnancy outcome, morbidity, and mortality of congenitally infected and non- infected newborns in bolivia. Am. J. Trop. Med. Hyg. 70, 201–209 (2004).
Torrico, F. et al. Are maternal re-
infections with Trypanosoma cruzi associated with higher morbidity and
mortality of congenital Chagas disease? Trop.
Med. Int. Heal. 11, 628–635 (2006).
Siriano, L. D. R., Luquetti, A. O.,
Avelar, J. B., Marra, N. L. & De Castro, A. M. Chagas disease: increased
parasitemia during pregnancy detected by
hemoculture. Am. J. Trop. Med. Hyg. 84, 569–574 (2011).
Brutus, L. et al. Short report: Detectable Trypanosoma cruzi parasitemia during pregnancy and delivery as a risk factor for congenital chagas disease. Am. J. Trop. Med. Hyg. 83, 1044–1047 (2010).
Duaso, J. et al. Trypanosoma cruzi induces apoptosis in ex vivo infected human chorionic villi. Placenta 32, 356–361 (2011).
Duaso, J. et al. Trypanosoma cruzi induces tissue disorganization and destruction of chorionic villi in an ex vivo infection model of human placenta. Placenta 31, 705–711 (2010).
Castillo, C. et al. Trypanosoma cruzi exosomes increases susceptibility to parasite infection in human placental chorionic villi explants. Placenta 51, 123–124 (2017).
Roberts, R. M., Green, J. A. & Schulz, L. C. The evolution of the placenta. Reproduction 152, R179–R189 (2016).
Turco, M. Y. et al. Trophoblast organoids as a model for maternal–fetal interactions during human placentation. Nature 564, 263–281 (2018).
Turco, M. Y. et al. Long- term,
hormone- responsive organoid cultures of human endometrium in a chemically
defined medium. Nat. Cell Biol. 19, 568–577 (2017).